X-AMR, a pop-up journal
Antimicrobial resistance (AMR) is a cross-disciplinary issue, with ground-breaking studies currently bringing together clinicians and modellers, veterinary and soil scientists, microbiologists and anthropologists. Yet finding a home for the unique publications from this research is difficult. The Microbiology Society is providing such a home with a new pop-up journal for cross-disciplinary research on antimicrobial resistance: X-AMR.
We invite submissions in the form of research papers, mini-reviews or commentaries. For more information on X-AMR, including how to submit your article, see our FAQs page.
Included in this collection are a host of antimicrobial resistance papers already published across our portfolio. The latest X-AMR articles will appear as and when they are published. Read our Guest Editors' introductory Editorial here.
Collection Contents
21 - 40 of 122 results
-
-
Rapid phenotypic evolution in multidrug-resistant Klebsiella pneumoniae hospital outbreak strains
More LessCarbapenem-resistant Klebsiella pneumoniae (CRKP) increasingly cause high-mortality outbreaks in hospital settings globally. Following a patient fatality at a hospital in Beijing due to a bla KPC-2-positive CRKP infection, close monitoring was put in place over the course of 14 months to characterize all bla KPC-2-positive CRKP in circulation in the hospital. Whole genome sequences were generated for 100 isolates from bla KPC-2-positive isolates from infected patients, carriers and the hospital environment. Phylogenetic analyses identified a closely related cluster of 82 sequence type 11 (ST11) isolates circulating in the hospital for at least a year prior to admission of the index patient. The majority of inferred transmissions for these isolates involved patients in intensive care units. Whilst the 82 ST11 isolates collected during the surveillance effort all had closely related chromosomes, we observed extensive diversity in their antimicrobial resistance (AMR) phenotypes. We were able to reconstruct the major genomic changes underpinning this variation in AMR profiles, including multiple gains and losses of entire plasmids and recombination events between plasmids, including transposition of bla KPC-2. We also identified specific cases where variation in plasmid copy number correlated with the level of phenotypic resistance to drugs, suggesting that the number of resistance elements carried by a strain may play a role in determining the level of AMR. Our findings highlight the epidemiological value of whole genome sequencing for investigating multi-drug-resistant hospital infections and illustrate that standard typing schemes cannot capture the extraordinarily fast genome evolution of CRKP isolates.
-
-
-
Very rapid flow cytometric assessment of antimicrobial susceptibility during the apparent lag phase of microbial (re)growth
More Less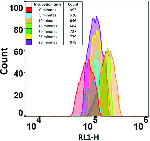 Rapid changes in the number and flow cytometric behaviour of cells of
E. coli
taken from a stationary phase and inoculated into rich medium.
Rapid changes in the number and flow cytometric behaviour of cells of
E. coli
taken from a stationary phase and inoculated into rich medium.Cells of E. coli were grown in LB medium, taken from a stationary phase of 2–4 h, and re-inoculated into fresh media at a concentration (105 ml−1 or lower) characteristic of bacteriuria. Flow cytometry was used to assess how quickly we could detect changes in cell size, number, membrane energization (using a carbocyanine dye) and DNA distribution. It transpired that while the lag phase observable macroscopically via bulk OD measurements could be as long as 4 h, the true lag phase could be less than 15–20 min, and was accompanied by many observable biochemical changes. Antibiotics to which the cells were sensitive affected these changes within 20 min of re-inoculation, providing the possibility of a very rapid antibiotic susceptibility test on a timescale compatible with a visit to a GP clinic. The strategy was applied successfully to genuine potential urinary tract infection (UTI) samples taken from a doctor’s surgery. The methods developed could prove of considerable value in ensuring the correct prescription and thereby lowering the spread of antimicrobial resistance.
-
-
-
Acquired qnrVC1 and bla NDM-1 resistance markers in an international high-risk Pseudomonas aeruginosa ST773 clone
More LessA multidrug-resistant Pseudomonas aeruginosa PS1 isolated from urine clinical sample was investigated in this study. The strain exhibited resistance to piperacillin/tazobactam, ciprofloxacin, imipenem, ceftazidime but it was susceptible to colistin. Analysis of whole-genome sequencing data by ResFinder detected various resistance determinants including qnrVC1 and bla NDM-1. The multiresistant P. aeruginosa isolate belonged to ST773 high-risk clone. The qnrVC1 and bla NDM-1 determinants were incorporated into different integrons. Expression of bla NDM-1 was fourfold and qnrVC1 was twofold increased, compared to that of rpsL housekeeping gene. Mutations in gyrA Thr83Leu and parC Ser87Leu were detected and additionally qnrVC1 expression indicates protective effect of QnrVC1 pentapeptid protein on gyrase and topoisomerase. High-risk P. aeruginosa clones integrate various carbapenemase and other resistance determinants into their genomes that facilitates further dissemination of multiresistance among clinical isolates. We report bla NDM-1 and qnrVC1 genes in P. aeruginosa ST773 international high-risk clone.
-
-
-
Antifungal susceptibilities, biofilms, phospholipase and proteinase activities in the Candida rugosa complex and Candida pararugosa isolated from tertiary teaching hospitals
More LessPurpose. Non-albicans Candida species have emerged as fungal pathogens that cause invasive infections, with many of these species displaying resistance to commonly used antifungal agents. This study was confined to studying the characteristics of clinical isolates of the C. rugosa complex and C. pararugosa species.
Methodology. Seven isolates of the C. rugosa complex and one isolate of C. pararugosa were obtained from two tertiary referral hospitals in Malaysia. Their antifungal susceptibilities, biofilm, proteinase, phospholipase, esterase and haemolysin activities were characterized. Biofilms were quantified using crystal violet (CV) and tetrazolium (XTT) reduction assays at 1.5, 6, 18, 24, 48 and 72 h.
Results/Key findings. The E-test antifungal tests showed that both species have elevated MICs compared to C. albicans and C. tropicalis. The highest biomass was observed in one of the C. rugosa isolates (0.237), followed by C. pararugosa (0.206) at 18 h of incubation. However, the highest bioactivity was observed in the C. rugosa ATCC 10571 strain at 24 h (0.075), followed by C. pararugosa at 48 h (0.048) and the same C. rugosa strain at 24 h (0.046), with P<0.05. All isolates exhibited high proteinase activity (+++) whereas six isolates showed very strong esterase activity (++++). All the isolates were alpha haemolytic producers. None of the isolates exhibited phospholipase activity.
Conclusion. Elevated MICs were shown for the C. rugosa complex and C. pararugosa for commonly used antifungal drugs. Further studies to identify virulence genes involved in the pathogenesis and genes that confer reduced drug susceptibility in these species are proposed.
-
-
-
Australian porcine clonal complex 10 (CC10) Escherichia coli belong to multiple sublineages of a highly diverse global CC10 phylogeny
More LessWe recently identified clonal complex 10 (CC10) Escherichia coli as the predominant clonal group in two populations of healthy Australian food-production pigs. CC10 are highly successful, colonizing humans, food-production animals, fresh produce and environmental niches. Furthermore, E. coli within CC10 are frequently drug resistant and increasingly reported as human and animal extra-intestinal pathogens. In order to develop a high-resolution global phylogeny and determine the repertoire of antimicrobial-resistance genes, virulence-associated genes and plasmid types within this clonal group, we downloaded 228 publicly available CC10 short-read genome sequences for comparison with 20 porcine CC10 we have previously described. Core genome single nucleotide polymorphism phylogeny revealed a highly diverse global phylogeny consisting of multiple lineages that did not cluster by geography or source of the isolates. Australian porcine strains belonged to several of these divergent lineages, indicative that CC10 is present in these animals due to multiple colonization events. Differences in resistance gene and plasmid carriage between porcine strains and the global collection highlighted the role of lateral gene transfer in the evolution of CC10 strains. Virulence profiles typical of extra-intestinal pathogenic E. coli were present in both Australian porcine strains and the broader collection. As both the core phylogeny and accessory gene characteristics appeared unrelated to the geography or source of the isolates, it is likely that the global expansion of CC10 is not a recent event and may be associated with faecal carriage in humans.
-
-
-
Genomic epidemiology of severe community-onset Acinetobacter baumannii infection
More LessAcinetobacter baumannii causes severe, fulminant, community-acquired pneumonia (CAP) in tropical and subtropical regions. We compared the population structure, virulence and antimicrobial resistance determinants of northern Australian community-onset A. baumannii strains with local and global strains. We performed whole-genome sequencing on 55 clinical and five throat colonization A. baumannii isolates collected in northern Australia between 1994 and 2016. Clinical isolates included CAP (n=41), healthcare-associated pneumonia (n=7) and nosocomial bloodstream (n=7) isolates. We also included 93 publicly available international A. baumannii genome sequences in the analyses. Patients with A. baumannii CAP were almost all critically unwell; 82 % required intensive care unit admission and 18 % died during their inpatient stay. Whole-genome phylogenetic analysis demonstrated that community-onset strains were not phylogenetically distinct from nosocomial strains. Some non-multidrug-resistant local strains were closely related to multidrug-resistant strains from geographically distant locations. Pasteur sequence type (ST)10 was the dominant ST and accounted for 31/60 (52 %) northern Australian strains; the remainder belonged to a diverse range of STs. The most recent common ancestor for ST10 was estimated to have occurred in 1738 (95 % highest posterior density, 1626–1826), with evidence of multiple introduction events between Australia and Southeast Asia between then and the present day. Virulence genes associated with biofilm formation and the type 6 secretion system (T6SS) were absent in many strains, and were not associated with in-hospital mortality. All strains were susceptible to gentamicin and meropenem; none carried an AbaR resistance island. Our results suggest that international dissemination of A. baumannii is occurring in the community on a contemporary timescale. Genes associated with biofilm formation and the T6SS may not be required for survival in community niches. The relative contributions of host and bacterial factors to the clinical severity of community-onset A. baumannii infection require further investigation.
-
-
-
Genotypic and phenotypic variations in methicillin-resistant Staphylococcus aureus isolates from outpatient, inpatient and nursing homes
More LessPurpose. The epidemiological shift in MRSA distribution from healthcare-related facilities to the general population is distressing and requires continuous monitoring to manage and control the rate of incidences.
Method. The retrospective relationship between genetic and phenotypic variability of methicillin-resistant Staphylococcus aureus (MRSA) isolates was determined in respect to the specimen source, patient location, sex and age. A total of 521 MRSA isolates were classified based on SCCmec, mec, agr, pvl and spa genetic markers using three different multiplex PCRs.
Results. Based on the genetic variability, the isolates were divided into 97 profiles, of which 59% belonged to only two profiles (P17 and P33). P17 was the predominate profile, harbouring SCCmecIVa, ccr2, mecB, agr1, spa413 and pvl markers. P17 was more prevalent among the younger population (average 33.9 years) from outpatient (77%) locations and wound (88%) sources. The second largest profile was P33, harbouring SCCmecII, ccr2+ccr3, mecA, agr2, spa413 and no PVL. P33 was more prevalent in the older population (average 70.7 years) and more common in females (62%) than males (38%). With respect to antibiotic resistance, P33 exhibited a high rate of resistance to penicillins, cephalosporins, fluoroquinolones and macrolides, and P17 had a lower resistance to fluoroquinolones.
Conclusion. This report contributes to the existing understanding of evolutionary epidemiology of antibiotic resistance in MRSA. The diversity of MRSA isolates and unique environmental preferences for each profile highlights the importance of epidemiological knowledge of MRSA distribution to determine the best treatment for patients in both community and hospital settings.
-
-
-
Oxidative stress response plays a role in antibiotic tolerance of Streptococcus mutans biofilms
More LessKnowledge about biofilm-associated antibiotic tolerance mechanisms is warranted in order to develop effective treatments against biofilm infections. We performed a screen of a Streptococcus mutans transposon mutant library for mutants with reduced biofilm-associated antimicrobial tolerance, and found that the spxA1 gene plays a role in tolerance towards gentamicin and other antibiotics such as vancomycin and linezolid. SpxA1 is a regulator of genes involved in the oxidative stress response in S. mutans . The oxidative stress response genes gor and ahpC were found to be up-regulated upon antibiotic treatment of S. mutans wild-type biofilms, but not spxA1 mutant biofilms. The gor gene product catalyses the formation of glutathione which functions as an important antioxidant during oxidative stress, and accordingly biofilm-associated antibiotic tolerance of the spxA1 mutant could be restored by exogenous addition of glutathione. Our results indicate that the oxidative stress response plays a role in biofilm-associated antibiotic tolerance of S. mutans , and add to the on-going debate on the role of reactive oxygen species in antibiotic mediated killing of bacteria.
-
-
-
Prevalence and genetic characterization of cephalosporin-resistant Enterobacteriaceae among dogs and cats in an animal shelter
More LessPurpose . The prevalence of antimicrobial-resistant bacteria, especially cephalosporin-resistant Enterobacteriaceae , is a major concern for human and animal health. We investigated the prevalence of cephalosporin-resistant Enterobacteriaceae among sheltered dogs and cats with various backgrounds.
Method . Faecal samples or rectal swabs were collected from 151 dogs and 182 cats, and screened for the presence of antimicrobial-resistant bacteria. Isolates were characterized phenotypically and genotypically by pulsed-field gel electrophoresis, multi-locus sequence typing and phylogenetic grouping. The animal attributes related to bacterial carriage were statistically analysed.
Results . Cephalosporin-resistant Enterobacteriaceae was detected in 22 dogs (14.6%) and 20 cats (11.0%): 21 were extended-spectrum β-lactamase (ESBL)-producing, 20 were AmpC-producing, and 1 was both ESBL- and AmpC-producing. Their β-lactamase genes were varied and associated with humans, animals or other origins. The genes CTX-M-14 (n=9) and CMY-2 (n=9) were dominant, but CTX-M-1, CTX-M-2, CTX-M-8, CTX-M-15, CTX-M-24, CTX-M-27, CTX-M-55 and DHA-1 genes were also detected. Genotyping of isolates revealed that β-lactamase-producing Enterobacteriaceae had high genetic diversity. Relationships between animals harbouring cephalosporin-resistant Enterobacteriaceae and individual attributes, such as sex and nutrition type, were detected, but there was no correlation between history of human association and the presence of the bacterium in either dogs or cats.
Conclusion . We found several types of cephalosporin-resistant Enterobacteriaceae distributed among companion animals with a range of individual attributes and histories in Osaka, Japan. Companion animals may play a bridging role in the circulation of antimicrobial-resistant bacteria from humans and from other origins.
-
-
-
Retrospective whole-genome sequencing analysis distinguished PFGE and drug-resistance-matched retail meat and clinical Salmonella isolates
More LessNon-typhoidal Salmonella is a leading cause of outbreak and sporadic-associated foodborne illnesses in the United States. These infections have been associated with a range of foods, including retail meats. Traditionally, pulsed-field gel electrophoresis (PFGE) and antibiotic susceptibility testing (AST) have been used to facilitate public health investigations of Salmonella infections. However, whole-genome sequencing (WGS) has emerged as an alternative tool that can be routinely implemented. To assess its potential in enhancing integrated surveillance in Pennsylvania, USA, WGS was used to directly compare the genetic characteristics of 7 retail meat and 43 clinical historic Salmonella isolates, subdivided into 3 subsets based on PFGE and AST results, to retrospectively resolve their genetic relatedness and identify antimicrobial resistance (AMR) determinants. Single nucleotide polymorphism (SNP) analyses revealed that the retail meat isolates within S. Heidelberg, S. Typhimurium var. O5- subset 1 and S. Typhimurium var. O5- subset 2 were separated from each primary PFGE pattern-matched clinical isolate by 6–12, 41–96 and 21–81 SNPs, respectively. Fifteen resistance genes were identified across all isolates, including fosA7, a gene only recently found in a limited number of Salmonella and a ≥95 % phenotype to genotype correlation was observed for all tested antimicrobials. Moreover, AMR was primarily plasmid-mediated in S. Heidelberg and S. Typhimurium var. O5- subset 2, whereas AMR was chromosomally carried in S. Typhimurium var. O5- subset 1. Similar plasmids were identified in both the retail meat and clinical isolates. Collectively, these data highlight the utility of WGS in retrospective analyses and enhancing integrated surveillance for Salmonella from multiple sources.
-
-
-
Synergistic activity of polymyxin B combined with vancomycin against carbapenem-resistant and polymyxin-resistant Acinetobacter baumannii: first in vitro study
More LessPurpose. The effect of a combination of polymyxin B (PMB) and vancomycin (VAN) was assessed against six Acinetobacter baumannii clinical isolates belonging to six different clusters (three PMB-susceptible and three PMB-resistant).
Methodology. The synergistic effect of the PMB–VAN combination was determined with the checkerboard, time-kill, disk-diffusion and M.I.C.Evaluator assays. PMB-resistance was investigated with mcr-1 gene amplification and a mutant frequency assay.
Results. In the checkerboard assay, all PMB-resistant isolates showed a synergistic effect. The time-kill assay demonstrated that the PMB–VAN combination had a bactericidal effect at 24 h against isolates with a high mutant rate for PMB, suggesting that this combination may block the hypermutation of some isolates. No antagonism was detected. All PMB-resistant isolates also showed synergism in the disk-diffusion test, and a significant decrease in VAN MICs in the M.I.C.Evaluator assay.
Conclusion. Our findings indicate that the PMB–VAN combination has a synergistic effect on A. baumannii , especially against PMB-resistant isolates.
-
-
-
The clinical significance of carbapenem-resistant Klebsiella pneumoniae rectal colonization in critically ill patients: from colonization to bloodstream infection
More LessPurpose . To highlight the clinical significance of carbapenem-resistant Klebsiella pneumoniae (CRKP) rectal colonization by examining the risk factors for CRKP rectal colonization and subsequent bloodstream infection (BSI) in critically ill patients.
Methodology . Prospective study of CRKP rectal colonization in an intensive care unit (ICU) during a 39-month period. CRKP strains isolated from both the blood cultures and corresponding rectal specimens (n=96) of patients were screened by PCR for the presence of antibiotic resistance-associated genes. Molecular analyses were conducted to investigate the clonal relatedness of CRKP strains from the rectal and blood specimens.
Results . Among the 498 patients, 226 were rectally colonized by CRKP, 48 of whom developed a CRKP BSI. The median time from hospital admission to the detection of CRKP rectal colonization was 8 days, while the median time from colonization to BSI was 4 days. The duration of ICU stay, patient/nurse ratio and prior use of antianaerobic antimicrobials were associated with CRKP rectal colonization. No specific factor was associated with BSIs in the colonized patients. The bla KPC-2 gene was detected in all 96 strains, which were all classified as sequence type ST-258. Representative pairs (n=48) of CRKP strains colonizing and infecting the same patient shared the same pulsotype.
Conclusion . Our results indicate that hospitalized patients become infected with their colonizing strains, supporting the strong association between colonization and BSI. Limiting antianaerobic antimicrobial administration, reducing the duration of ICU stay and maintaining a low patient/nurse ratio are possible strategies to restrict rectal CRKP colonization in ICUs.
-
-
-
Utilizing genomic analyses to investigate the first outbreak of van A vancomycin-resistant Enterococcus in Australia with emergence of daptomycin non-susceptibility
More LessIntroduction. The majority of vancomycin-resistant Enterococcus faecium (VREfm) in Australia is of the vanB genotype. An outbreak of vanA VREfm emerged in our haematology/oncology unit between November 2014 and May 2015. The first case of daptomycin non-susceptible E. faecium (DNSEfm) detected was a patient with vanA VREfm bacteraemia who showed clinical failure of daptomycin therapy, prompting microbiologic testing confirming daptomycin non-susceptibility.
Objectives. To describe the patient profiles, antibiotic susceptibility and genetic relatedness of vanA VREfm isolates in the outbreak.
Methods. Chart review of vanA VREfm colonized and infected patients was undertaken to describe the demographics, clinical features and outcomes of therapy. Whole genome sequencing of vanA VREfm isolates involved in the outbreak was conducted to assess clonality.
Results. In total, 29 samples from 24 patients tested positive for vanA VREfm (21 screening swabs and 8 clinical isolates). Five isolates were DNSEfm (four patients colonized, one patient with bacteraemia), with only one patient exposed to daptomycin previously. In silico multi-locus sequence typing of the isolates identified 25/26 as ST203, and 1/26 as ST796. Comparative genomic analysis revealed limited core genome diversity amongst the ST203 isolates, consistent with an outbreak of a single clone of vanA VREfm.
Conclusions. Here we describe an outbreak of vanA VREfm in a haematology/oncology unit. Genomic analysis supports transmission of an ST203 vanA VRE clone within this unit. Daptomycin non-susceptibility in 5/24 patients left linezolid as the only treatment option. Daptomycin susceptibility cannot be assumed in vanA VREfm isolates and confirmatory testing is recommended.
-
-
-
An improved carbapenem inactivation method, CIMTrisII, for carbapenemase production by Gram-negative pathogens
More LessThe modified carbapenem inactivation method (mCIM) is a simple phenotypic screening method for detecting carbapenemase production by Enterobacteriaceae and Pseudomonas aeruginosa . We recently developed another modified carbapenem inactivation method (CIMTris), in which carbapenemase is extracted from bacteria with Tris-HCl buffer, to detect carbapenemase production by Acinetobacter and Pseudomonas species. This study describes an improved carbapenem inactivation method, CIMTrisII, for detecting carbapenemase production by Gram-negative pathogens, including Enterobacteriaceae , Acinetobacter and Pseudomonas species. CIMTrisII was different from CIMTris in the concentration of Meropenem disks (5-µg MEM disks vs. 10-µg MEM disks), the inoculum volume of the bacteria (a 5-µl loopful vs. a 10 µl loopful) and the incubation time (1 vs. 2 h). CIMTrisII showed an overall sensitivity of 99.3 % and an overall specificity of 95.0 % for tested isolates. In comparison, CIMTris showed a sensitivity of 96.1 % and a specificity of 96.3 %, and mCIM showed a sensitivity of 67.1 % and a specificity of 100 %. CIMTrisII is thus deemed useful for detecting carbapenemase production by Gram-negative pathogens.
-
-
-
Antibiotic resistance of Campylobacter jejuni isolates recovered from humans with diarrhoea in Turkey
More LessPurpose. This study was aimed at investigating the occurrence and genetic mechanisms of resistance to ciprofloxacin, tetracycline and erythromycin in clinical isolates of Campylobacter jejuni recovered from human cases of acute gastroenteritis in Turkey.
Methodology. MIC values of each antibiotic were determined with the epsilometer test (E-test). Resistance genes/mutations were first screened by PCR and analysed by subsequent DNA sequencing.
Results. From a total of 152 C. jejuni isolates tested, 113 (74.3%), 38 (25%) and 9 (5.9%) were found to be resistant to ciprofloxacin, tetracycline and erythromycin, respectively. Sequence analysis of ciprofloxacin-resistant isolates showed that all resistant strains (n=113) carried Thr-86-Ile substition in the gyrA gene, which is the most frequently observed mutation in fluoroquinolone-resistant Campylobacter . All of the tetracycline-resistant isolates (n=38) carried the tetO gene. All of the erythromycin-resistant isolates (n=9) harboured the point mutation A2075G in the 23S rRNA gene, which is the most common mutation conferring macrolide resistance in C. jejuni .
Conclusion. The phenotypic susceptibility testing results were found to agree well with those obtained by genetic detection methods for the C. jejuni isolates tested. The findings of this study showed a very high level of resistance to ciprofloxacin and to a lesser extent to tetracycline while resistance to erythromycin remained at a low level. Thus, erythromycin may be considered as the first choice for treatment of Campylobacter infections in this geographical region when indicated.
-
-
-
How actively should we screen for chlamydia and gonorrhoea in MSM and other high-ST-prevalence populations as we enter the era of increasingly untreatable infections? A viewpoint
More LessA number of national and international organizations are advocating more intensive screening for Chlamydia trachomatis and Neisseria gonorrhoeae in high-prevalence populations as a way to reduce the prevalence of these infections. In this article, we review the available evidence and conclude that there is a paucity of evidence to support this approach. We further hypothesize that increasing screening intensity in high-prevalence populations will result in a considerable risk for the emergence of antimicrobial resistance in Neisseria gonorrhoeae and other pathobionts.
-
-
-
Prevalence of macrolide resistance in Treponema pallidum is associated with macrolide consumption
More LessWe hypothesized that the large global variations in the prevalence of macrolide resistance in Treponema pallidum are related to differences in population-level macrolide consumption. The hypothesis was tested by, at a country-level, regressing the peak prevalence of macrolide resistance against the national macrolide consumption in the year prior to this, controlling for the year of the resistance prevalence estimate. A strong association was found between the per capita consumption of macrolides and macrolide resistance (coefficient 0.7, 95 % confidence interval 0.2–0.12, P=0.009).
-
-
-
Evolution of a clade of Acinetobacter baumannii global clone 1, lineage 1 via acquisition of carbapenem- and aminoglycoside-resistance genes and dispersion of ISAba1
More LessResistance to carbapenem and aminoglycoside antibiotics is a critical problem in Acinetobacter baumannii , particularly when genes conferring resistance are acquired by multiply or extensively resistant members of successful globally distributed clonal complexes, such as global clone 1 (GC1) . Here, we investigate the evolution of an expanding clade of lineage 1 of the GC1 complex via repeated acquisition of carbapenem- and aminoglycoside-resistance genes. Lineage 1 arose in the late 1970s and the Tn6168/OCL3 clade arose in the late 1990s from an ancestor that had already acquired resistance to third-generation cephalosporins and fluoroquinolones. Between 2000 and 2002, two distinct subclades have emerged, and they are distinguishable via the presence of an integrated phage genome in subclade 1 and AbaR4 (carrying the oxa23 carbapenem-resistance gene in Tn2006) at a specific chromosomal location in subclade 2. Part or all of the original resistance gene cluster in the chromosomally located AbaR3 has been lost from some isolates, but plasmids carrying alternate resistance genes have been gained. In one group in subclade 2, the chromosomally located AbGRI3, carrying the armA aminoglycoside-resistance gene, has been acquired from a GC2 isolate and incorporated via homologous recombination. ISAba1 entered the common ancestor of this clade as part of the cephalosporin-resistance transposon Tn6168 and has dispersed differently in each subclade. Members of subclade 1 share an ISAba1 in one specific position in the chromosome and in subclade 2 two different ISAba1 locations are shared. Further shared ISAba1 locations distinguish further divisions, potentially providing simple markers for epidemiological studies.
-
-
-
Fosfomycin: mechanisms and the increasing prevalence of resistance
More LessThere are challenges regarding increased global rates of microbial resistance and the emergence of new mechanisms that result in microorganisms becoming resistant to antimicrobial drugs. Fosfomycin is a broad-spectrum bactericidal antibiotic effective against Gram-negative and certain Gram-positive bacteria, such as Staphylococci, that interfere with cell wall synthesis. During the last 40 years, fosfomycin has been evaluated in a wide range of applications and fields. Although numerous studies have been done in this area, there remains limited information regarding the prevalence of resistance. Therefore, in this review, we focus on the available data concerning the mechanisms and increasing resistance regarding fosfomycin.
-
-
-
Multi-step genomic dissection of a suspected intra-hospital Helicobacter cinaedi outbreak
More LessHelicobacter cinaedi is an emerging pathogen causing bacteraemia and cellulitis. Nosocomial transmission of this microbe has been described, but detailed molecular-epidemiological analyses have not been performed. Here, we describe the results of a multi-step genome-wide phylogenetic analysis of a suspected intra-hospital outbreak of H. cinaedi that occurred in a hospital in Japan. The outbreak was recognized by the infectious control team (ICT) of the hospital as a sudden increase in H. cinaedi bacteraemia. ICT defined this outbreak case based on 16S rRNA sequence data and epidemiological information, but were unable to determine the source and route of the infections. We therefore re-investigated this case using whole-genome sequencing (WGS). We first performed a species-wide analysis using publicly available genome sequences to understand the level of genomic diversity of this under-studied species. The clusters identified were then separately analysed using the genome sequence of a representative strain in each cluster as a reference. These analyses provided a high-level phylogenetic resolution of each cluster, identified a confident set of outbreak isolates, and discriminated them from other closely related but distinct clones, which were locally circulating and invaded the hospital during the same period. By considering the epidemiological data, possible strain transmission chains were inferred, which highlighted the role of asymptomatic carriers or environmental contamination. The emergence of a subclone with increased resistance to fluoroquinolones in the outbreak was also recognized. Our results demonstrate the impact of the use of a closely related genome as a reference to maximize the power of WGS.
-