X-AMR, a pop-up journal
Antimicrobial resistance (AMR) is a cross-disciplinary issue, with ground-breaking studies currently bringing together clinicians and modellers, veterinary and soil scientists, microbiologists and anthropologists. Yet finding a home for the unique publications from this research is difficult. The Microbiology Society is providing such a home with a new pop-up journal for cross-disciplinary research on antimicrobial resistance: X-AMR.
We invite submissions in the form of research papers, mini-reviews or commentaries. For more information on X-AMR, including how to submit your article, see our FAQs page.
Included in this collection are a host of antimicrobial resistance papers already published across our portfolio. The latest X-AMR articles will appear as and when they are published. Read our Guest Editors' introductory Editorial here.
Collection Contents
1 - 100 of 122 results
-
-
Expression of adhesin genes and biofilm formation among Klebsiella oxytoca clinical isolates from patients with antibiotic-associated haemorrhagic colitis
More LessPurpose. Biofilm formation and resistance to last-line antibiotics have restricted chemotherapy options toward infection eradication.
Methodology. Fifty K. oxytoca isolates were collected from patients with antibiotic-associated haemorrhagic colitis (AAHC). Antibiotic susceptibility tests were conducted and phenotypic biofilm formation was assessed using microtitre tissue plate (MTP) assay. PCR was employed to amplify the adhesins, extended-spectrum β-lactamases (ESBLs), carbapenemase and colistin resistance genes. The expression of adhesin genes was evaluated using quantitative real-time PCR (RT-qPCR).
Results/Key findings. The previous antibiotic consumption and hospitalization (P<0.05) and older ages (P=0.0033) were significantly associated with AAHC. None of the isolates produced biofilm strongly, but 70% of them produced moderate-level biofilm. The bla CTX-M (12/14), the bla IMP (8/14 MICIMI =4 µg ml−1 ) and bla OXA-48-like (5/14) and mcr-1 (4/14) genes were predominant, three of which harbouring all the genes. The expression of matB (0.023) and mrkA (0.011) was significantly different between multidrug-resistant and susceptible isolates. Furthermore, moderately biofilm producer isolates significantly exhibited higher expression of fimA (P=.0117), pilQ (P=0.002) and mrkA (P=0.020) genes compared to biofilm non-producers. No significant difference regarding gene expression was observed among ESBL alleles.
Conclusion. Bacterial attachment by adhesins and biofilm formation among extensive drug-resistant K. oxytoca isolates hinder the efficient infection eradication. Hence, control and surveillance studies should be performed and other therapeutic auspicious approaches must be taken into account against AAHC, biofilm formation and drug resistance spread. Furthermore, previous antibiotic consumption and long-term hospitalization should be controlled.
-
-
-
In vitro activity of mecillinam and nitroxoline against Neisseria gonorrhoeae – re-purposing old antibiotics in the multi-drug resistance era
More LessIn 2018, the European Centre for Disease Prevention and Control reported the first cases of extensively drug-resistant Neisseria gonorrhoeae infections in Europe. Seeking new options for antimicrobial therapy we investigated the susceptibility of N. gonorrhoeae to nitroxoline (NIT) and mecillinam (MCM), both of which are currently only indicated to treat uncomplicated urinary tract infections. Clinical N. gonorrhoeae isolates with non-susceptibility to penicillin from two German medical centres were included (n =27). Most isolates were also non-susceptible to a range of other anti-gonococcal antimicrobials (cefotaxime, ciprofloxacin, azithromycin, tetracycline). All isolates were further characterized by multi-locus sequence typing. MICs of penicillin and cefotaxime were determined by agar gradient diffusion. Production of penicillinase was tested by cefinase disk test. Susceptibility of MCM was investigated by agar dilution, NIT by agar dilution and disk diffusion. Penicillin MICs ranged from 0.125 to 64 mg l−1 and MICs of cefotaxime ranged from < 0.016 to 1 mg l−1 . Five isolates were penicillinase-producers. MICs of MCM ranged from 16 to > 128 mg l−1 whereas MICs of NIT ranged from 0.125 to 2 mg l−1 . NIT disk diffusion (median zone diameter 32 mm) correlated well with results from agar dilution. We demonstrated excellent in vitro activity of NIT against clinical N. gonorrhoeae isolates with non-susceptibility to standard anti-gonococcal antibiotics. MCM activity was unsatisfactory. Correlation of agar dilution and disk diffusion in NIT susceptibility testing is an important aspect with potential clinical implications.
-
-
-
Multidrug- and colistin-resistant Salmonella enterica 4,[5],12:i:- sequence type 34 carrying the mcr-3.1 gene on the IncHI2 plasmid recovered from a human
More LessA colistin-resistant Salmonella enterica 4, [5],12:i:- sequence type (ST) 34 harbouring mcr-3.1 was recovered from a patient who travelled to China 2 weeks prior to diarrhoea onset. Genomic analysis revealed the presence of the mcr-3.1 gene located in the globally disseminated IncHI2 plasmid, highlighting the intercontinental dissemination of the colistin-resistant S. enterica 4, [5],12:i:- ST34 pandemic clone.
-
-
-
Nisin penetration and efficacy against Staphylococcus aureus biofilms under continuous-flow conditions
More LessBiofilms may enhance the tolerance of bacterial pathogens to disinfectants, biocides and other stressors by restricting the penetration of antimicrobials into the matrix-enclosed cell aggregates, which contributes to the recalcitrance of biofilm-associated infections. In this work, we performed real-time monitoring of the penetration of nisin into the interior of Staphylococcus aureus biofilms under continuous flow and compared the efficacy of this lantibiotic against planktonic and sessile cells of S. aureus . Biofilms were grown in Center for Disease Control (CDC) reactors and the spatial and temporal effects of nisin action on S. aureus cells were monitored by real-time confocal microscopy. Under continuous flow, nisin caused loss of membrane integrity of sessile cells and reached the bottom of the biofilms within ~20 min of exposure. Viability analysis using propidium iodide staining indicated that nisin was bactericidal against S. aureus biofilm cells. Time-kill assays showed that S. aureus viability reduced 6.71 and 1.64 log c.f.u. ml-1 for homogenized planktonic cells in exponential and stationary phase, respectively. For the homogenized and intact S. aureus CDC biofilms, mean viability decreased 1.25 and 0.50 log c.f.u. ml-1, respectively. Our results demonstrate the kinetics of biofilm killing by nisin under continuous-flow conditions, and shows that alterations in the physiology of S. aureus cells contribute to variations in sensitivity to the lantibiotic. The approach developed here could be useful to evaluate the antibiofilm efficacy of other bacteriocins either independently or in combination with other antimicrobials.
-
-
-
Rapid antimicrobial susceptibility tests for sepsis; the road ahead
More LessCurrent methods for antimicrobial susceptibility testing (AST) are too slow to affect initial treatment decisions in the early stages of sepsis, when the prescriber is most concerned to select effective therapy immediately, rather than finding out what will not work 1 or 2 days later. There is a clear need for much faster differentiation between viral and bacterial infection, and AST, linked to earlier aetiological diagnosis, without sacrificing either the accuracy of quantitative AST or the low cost of qualitative AST. Truly rapid AST methods are eagerly awaited, and there are several candidate technologies that aim to improve the targeting of our limited stock of effective antimicrobial agents. However, none of these technologies are approaching the point of care and nor can they be described as truly culture-independent diagnostic tests. Rapid chemical and genomic methods of resistance detection are not yet reliable predictors of antimicrobial susceptibility and often rely on prior bacterial isolation. In order to resolve the trade-off between diagnostic confidence and therapeutic efficacy in increasingly antimicrobial-resistant sepsis, we propose a series of three linked decision milestones: initial clinical assessment (e.g. qSOFA score) within 10 min, initial laboratory tests and presumptive antimicrobial therapy within 1 h, and definitive AST with corresponding antimicrobial amendment within an 8 h window (i.e. the same working day). Truly rapid AST methods therefore must be integrated into the clinical laboratory workflow to ensure maximum impact on clinical outcomes of sepsis, and diagnostic and antimicrobial stewardship. The requisite series of development stages come with a substantial regulatory burden that hinders the translation of innovation into practice. The regulatory hurdles for the adoption of rapid AST technology emphasize technical accuracy, but progress will also rely on the effect rapid AST has on prescribing behaviour by physicians managing the care of patients with sepsis. Early adopters in well-equipped teaching centres in close proximity to large clinical laboratories are likely to be early beneficiaries of rapid AST, while simplified and lower-cost technology is needed to support poorly resourced hospitals in developing countries, with their higher burden of AMR. If we really want the clinical laboratory to deliver a specific, same-day diagnosis underpinned by definitive AST results, we are going to have to advocate more effectively for the clinical benefits of bacterial detection and susceptibility testing at critical decision points in the sepsis management pathway.
-
-
-
Circulation of imipenem-resistant Acinetobacter baumannii ST10, ST2 and ST3 in a university teaching hospital from Tehran, Iran
More LessPurpose. Multi-drug resistant (MDR) Acinetobacter baumannii has introduced a worldwide health crisis. The purposes of this study were to characterize the clonal relatedness among MDR clinical strains and to introduce a new two-locus typing method confirmed by multi-locus sequence typing (MLST).
Methodology. In this study, we determined antimicrobial resistance, detected genes associated with carbapenem resistance and characterized clonal relatedness among 99 clinical isolates extracted from 82 hospitalized inpatients in a university hospital.
Results. Of the 99 A. baumannii isolates, 92.9% (92/99) were resistant to imipenem and 97.9% (97/99) had an MDR profile. We found that the high prevalence of blaVIM [94.9% (94/99)] and blaOXA-23-like [93.93% (93/99)] is the main mechanism of carbapenem resistance. This study proposes a new two-locus typing (blaOXA-51-like and ampC) method for the rapid identification of clonal complexes (CCs). The results of this method and confirmation by MLST show that clinical isolates carry blaOXA-68 as well as ampC-10 or ampC-20 genes belonging to CC10 (ST10); blaOXA-66 and ampC-2 belonging to CC2 (ST2); and blaOXA-71 and ampC-3 belonging to CC3 (ST3). One isolate had blaOXA-90 with an undetermined allele number of ampC belonging to ST513.
Conclusion. The high prevalence of MDR strains and the circulation of four limited clones, including ST10 (45/99), ST2 (41/99), ST3 (12/99) and ST513 (1/99), in the clinical setting highlights the importance of a rigorous infection control programme. The two-locus typing method has more discrimination than the application of each method separately and it could be applied for the rapid determination of the CC without performing MLST.
-
-
-
Erythromycin-resistant Streptococcus pneumoniae: phenotypes, genotypes, transposons and pneumococcal vaccine coverage rates
More LessPurpose. To assess the antibiotic resistance, transposon profiles, serotype distribution and vaccine coverage rates in 110 erythromycin-resistant S. pneumoniae clinical isolates.
Methodology. Erythromycin, clindamycin, tetracycline, chloramphenicol and kanamycin susceptibilities were assessed using the E-test/disc diffusion method. Inducible macrolide resistance was tested using the erythromycin-clindamycin double disc diffusion test. Serogrouping and serotyping were performed using latex particle agglutination and the Quellung reaction, respectively. Drug resistance genes and transposon-specific genes were investigated by PCR.
Results. Of the isolates, 93 % were resistant to clindamycin; 81 % were resistant to tetracycline; 76 % were multi-drug-resistant, having resistance to both clindamycin and tetracycline; and 12 % had extended-drug resistance, being resistant to clindamycin, tetracycline, chloramphenicol and kanamycin. The majority of isolates (88.2 %) exhibited the cMLSB phenotype. The association between the cMLSB phenotype and tetracycline resistance was related to transposons Tn2010 (38.2 %), Tn6002 (21.8 %) and Tn3872 (18.2 %). M and iMLSB phenotypes were observed in 7 and 5 % of the isolates, respectively. The most frequent serotype was 19 F (40 %). Among the erythromycin-resistant pneumococci, vaccine coverage rates for the 13-valent pneumococcal conjugate vaccine (PCV-13) and the 23-valent pneumococcal polysaccharide vaccine (PPSV-23) were 76.4 and 79.1 %, respectively, compared to 82.2 and 85.1 % transposon-carrying isolates.
Conclusions. Multi-drug resistance among erythromycin-resistant S. pneumoniae isolates mainly occurs due to the horizontal spread of the Tn916 family of transposons. The majority of the transposon-carrying isolates are covered by 13- and 23-valent pneumococcal vaccines. Since serotype distribution and transposons in S. pneumoniae isolates may change over time, close monitoring is essential.
-
-
-
Evaluation of risk factors for colistin resistance among uropathogenic isolates of Escherichia coli and Klebsiella pneumoniae: a case–control study
More LessIntroduction. The last few years have seen the emergence of multi-drug resistant (MDR) Gram-negative infections, which are associated with high morbidity and mortality. The indiscriminate use of colistin has led to the development of resistance, which can be diagnosed effectively by broth microdilution. Studies from India are limited, and this study was conducted in order to determine the prevalence and risk factors associated with colistin resistance.
Methods. Urine samples from patients admitted with urinary tract infection (UTI), growing MDR Escherichia coli and Klebsiella pneumoniae , were tested for the minimum inhibitory concentration (MIC) of colistin by broth microdilution. Isolates with an MIC >2 µg ml−1 (resistant) were subjected to polymerase chain reaction (PCR) for the mcr1, mcr2 and mgrB genes. A case–control study with 21 cases (resistant) and 42 matched controls (sensitive) was designed to evaluate risk factors and outcomes (recurrent UTI, readmission and hospital stay >2 weeks).
Results. Two hundred and fifty MDR isolates ( E. coli =142/2319 and K.pneumoniae=108/775) from 216 patients were selected from the 25 046 isolates screened. Twenty-five isolates (20 K.pneumoniae and 5 E. coli ) were resistant to colistin, with a prevalence of 3.52 % in E. coli and 18.5 % in K. pneumoniae among the MDR isolates. PCR for the mcr1 and mcr2 genes was negative. Multivariate regression showed that multiple episodes of hospitalization, hospital stay >2 weeks, exposure to >three antibiotic classes and abnormality/surgery of the lower urinary tract were the significant risk factors for colistin resistance. Previous use of colistin and colistin resistance had a significant effect on all outcomes.
Conclusions. K. pneumoniae show six times higher prevalence of colistin resistance than E. coli , and the emergence of resistant organisms has led to an increase in morbidity in infected patients.
-
-
-
Heterogeneity of ROS levels in antibiotic-exposed mycobacterial subpopulations confers differential susceptibility
More LessPhenotypically heterogeneous but genetically identical mycobacterial subpopulations exist in in vitro cultures, in vitro-infected macrophages, infected animal models and tuberculosis patients. In this regard, we recently reported the presence of two subpopulations of cells, which are phenotypically different in length and buoyant density, in mycobacterial cultures. These are the low-buoyant-density short-sized cells (SCs), which constitute ~10–20 % of the population, and the high-buoyant-density normal/long-sized cells (NCs), which form ~80–90 % of the population. The SCs were found to be significantly more susceptible to rifampicin (RIF), isoniazid (INH), H2O2 and acidified nitrite than the NCs. Here we report that the RIF-/INH-/H2O2-exposed SCs showed significantly higher levels of oxidative stress and therefore higher susceptibility than the equivalent number of exposed NCs. Significantly higher levels of hydroxyl radical and superoxide were found in the antibiotic-exposed SCs than in the equivalently exposed NCs. Different proportions of the subpopulation of SCs were found to have different levels of reactive oxygen species (ROS). The hydroxyl radical quencher, thiourea, and the superoxide dismutase mimic, TEMPOL, significantly reduced hydroxyl radical and superoxide levels, respectively, in the antibiotic-exposed SCs and NCs and thereby decreased their differential susceptibility to antibiotics. Thus, the present study shows that the heterogeneity of the reactive oxygen species (ROS) levels in these mycobacterial subpopulations confers differential susceptibility to antibiotics. We have discussed the possible mechanisms that can generate differential ROS levels in the antibiotic-exposed SCs and NCs. The present study advances our current understanding of the molecular mechanisms underlying antibiotic tolerance in mycobacteria.
-
-
-
Reduced vancomycin susceptibility and increased macrophage survival in Staphylococcus aureus strains sequentially isolated from a bacteraemic patient during a short course of antibiotic therapy
More LessPurpose. The purpose of the present study was to determine the relatedness of Staphylococcus aureus strains successively isolated over a 7-day period from a single bacteraemic patient undergoing antibiotic treatment with vancomycin.
Methods. The S. aureus strains had been isolated and sequenced previously. Antibiotic susceptibility testing, population analysis profiling, and lysostaphin sensitivity and phagocytic killing assays were used to characterize these clonal isolates.
Results. The seven isolates (MEH1–MEH7) were determined to belong to a common multilocus sequence type (MLST) and spa type. Within the third and fifth day of vancomycin treatment, mutations were observed in the vraS and rpsU genes, respectively. Population analysis profiles revealed that the initial isolate (MEH1) was vancomycin-susceptible S. aureus (VSSA), while those isolated on day 7 were mostly heteroresistant vancomycin-intermediate S. aureus (hVISA). Supporting these findings, MEH7 was also observed to be slower in growth, to have an increase in cell wall width and to have reduced sensitivity to lysostaphin, all characteristics of VISA and hVISA strains. In addition, MEH7, although phagocytosed at numbers comparable to the initial isolate, MEH1, survived in higher numbers in RAW 264.7 macrophages. Macrophages infected with MEH7 also released more TNF-α and IFN-1β.
Conclusion. We report an increasing resistance to vancomycin coupled with daptomycin that occurred within approximately 3 days of receiving vancomycin and steadily increased until the infection was cleared with an alternative antibiotic therapy. This study reiterates the need for rapid, efficient and accurate detection of hVISA and VISA infections, especially in high-bacterial load, metastatic infections like bacteraemia.
-
-
-
Whole genome sequencing of NDM-1-producing serotype K1 ST23 hypervirulent Klebsiella pneumoniae in China
More LessBao-Tao Liu and Wei-Qi SuPurpose. The emergence and spread of carbapenem-resistant hypervirulent Klebsiella pneumoniae (CR-hvKP) is causing worldwide concern, whereas NDM-producing hvKP is still rare. Here we report the complete genome sequence characteristics of an NDM-1-producing ST23 type clinical hvKP in PR China.
Methodology. Capsular polysaccharide serotyping was performed by PCR. The complete genome sequence of isolate 3214 was obtained using both the Illumina Hiseq platform and Pacbio RS platform. Multilocus sequence type was identified by submitting the genome sequence to mlst 2.0 and the antimicrobial resistance genes and plasmid replicons were identified using ResFinder and PlasmidFinder, respectively. Transferability of the bla NDM-1-bearing plasmid was determined by conjugation experiment, S1 pulsed-field gel electrophoresis and Southern hybridization.
Results. Isolate 3214 was classified to ST23 and belonged to the K1 capsular serotype. The isolate’s total genome size was 6 171 644 bp with a G+C content of 56.39 %, consisting of a 5 448 209 bp chromosome and seven plasmids. The resistome included 18 types of antibiotic resistance genes. Fourteen resistance genes including bla NDM-1 and bla CTX-M-14 were located on plasmids and five also including bla CTX-M-14 were in the chromosome. Plasmid pNDM_3214 carrying bla NDM-1 harboured six types of resistance genes surrounded by insertion sequences and was conjugative. The worldwide pLVPK-like virulence plasmid harbouring rmpA2 and rmpA was also found in this isolate.
Conclusion. This study provides basic information of phenotypic and genomic features of ST23 CR-hvKP isolate 3214. Our data highlights the potential risk of spread of NDM-1-producing ST23 hvKP.
-
-
-
Antibiotic resistomes of healthy pig faecal metagenomes
More LessAntibiotic resistance reservoirs within food-producing animals are thought to be a risk to animal and human health. This study describes the minimum natural resistome of pig faeces as the bacteria are under no direct antibiotic selective pressure. The faecal resistome of 257 different genes comprised 56 core and 201 accessory resistance genes. The genes present at the highest relative abundances across all samples were tetW, tetQ, tet44, tet37, tet40, mefA, aadE, ant(9)−1, ermB and cfxA2. This study characterized the baseline resistome, the microbiome composition and the metabolic components described by the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways in healthy pig faeces, without antibiotic selective pressures. The microbiome hierarchical analysis resulted in a cluster tree with a highly similar pattern to that of the accessory resistome cluster tree. Functional capacity profiling identified genes associated with horizontal gene transfer. We identified a statistically significant positive correlation between the total antibiotic resistome and suggested indicator genes, which agree with using these genes as indicators of the total resistomes. The correlation between total resistome and total microbiome in this study was positive and statistically significant. Therefore, the microbiome composition influenced the resistome composition. This study identified a core and accessory resistome present in a cohort of healthy pigs, in the same conditions without antibiotics. It highlights the presence of antibiotic resistance in the absence of antibiotic selective pressure and the variability between animals even under the same housing, food and living conditions. Antibiotic resistance will remain in the healthy pig gut even when antibiotics are not used. Therefore, the risk of antibiotic resistance transfer from animal faeces to human pathogens or the environment will remain in the absence of antibiotics.
-
-
-
Caribbean multi-centre study of Klebsiella pneumoniae: whole-genome sequencing, antimicrobial resistance and virulence factors
More LessThe surveillance of antimicrobial-resistant isolates has proven to be one of the most valuable tools to understand the global rise of multidrug-resistant bacterial pathogens. We report the first insights into the current situation in the Caribbean, where a pilot project to monitor antimicrobial resistance (AMR) through phenotypic resistance measurements combined with whole-genome sequencing was set up in collaboration with the Caribbean Public Health Agency (CARPHA). Our first study focused on Klebsiella pneumoniae , a highly relevant organism amongst the Gram-negative opportunistic pathogens worldwide causing hospital- and community-acquired infections. Our results show that not only carbapenem resistance, but also hypervirulent strains, are circulating in patients in the Caribbean. Our current data does not allow us to infer their prevalence in the population. We argue for the urgent need to further support AMR surveillance and stewardship in this almost uncharted territory, which can make a significant impact on the reduction of antimicrobial usage. This article contains data hosted by Microreact (https://microreact.org).
-
-
-
Genomic profile of Brazilian methicillin-resistant Staphylococcus aureus resembles clones dispersed worldwide
More LessPurpose. Comparative genomic analysis of strains may help us to better understand the wide diversity of their genetic profiles. The aim of this study was to analyse the genomic features of the resistome and virulome of Brazilian first methicillin-resistant Staphylococcus aureus (MRSA) isolates and their relationship to other Brazilian and international MRSA strains.
Methodology. The whole genomes of three MRSA strains previously isolated in Vitória da Conquista were sequenced, assembled, annotated and compared with other MRSA genomes. A phylogenetic tree was constructed and the pan-genome and accessory and core genomes were constructed. The resistomes and virulomes of all strains were identified.
Results/Key findings. Phylogenetic analysis of all 49 strains indicated different clones showing high similarity. The pan-genome of the analysed strains consisted of 4484 genes, with 31 % comprising the gene portion of the core genome, 47 % comprising the accessory genome and 22 % being singletons. Most strains showed at least one gene related to virulence factors associated with immune system evasion, followed by enterotoxins. The strains showed multiresistance, with the most recurrent genes conferring resistance to beta-lactams, fluoroquinolones, aminoglycosides and macrolides.
Conclusions. Our comparative genomic analysis showed that there is no pattern of virulence gene distribution among the clones analysed in the different regions. The Brazilian strains showed similarity with clones from several continents.
-
-
-
Genomic surveillance of Escherichia coli in municipal wastewater treatment plants as an indicator of clinically relevant pathogens and their resistance genes
More LessWe examined whether genomic surveillance of Escherichia coli in wastewater could capture the dominant E. coli lineages associated with bloodstream infection and livestock in the East of England, together with the antibiotic-resistance genes circulating in the wider E. coli population. Treated and untreated wastewater was taken from 20 municipal treatment plants in the East of England, half in direct receipt of acute hospital waste. All samples were culture positive for E. coli , and all but one were positive for extended-spectrum β-lactamase (ESBL)-producing E. coli . The most stringent wastewater treatment (tertiary including UV light) did not eradicate ESBL- E. coli in 2/3 cases. We sequenced 388 E. coli (192 ESBL, 196 non-ESBL). Multilocus sequence type (ST) diversity was similar between plants in direct receipt of hospital waste versus the remainder (93 vs 95 STs, respectively). We compared the genomes of wastewater E. coli with isolates from bloodstream infection (n=437), and livestock farms and retail meat (n=431) in the East of England. A total of 19/20 wastewater plants contained one or more of the three most common STs associated with bloodstream infection (ST131, ST73, ST95), and 14/20 contained the most common livestock ST (ST10). In an analysis of 1254 genomes (2 cryptic E. coli were excluded), wastewater isolates were distributed across the phylogeny and intermixed with isolates from humans and livestock. Ten bla CTX-M elements were identified in E. coli isolated from wastewater, together with a further 47 genes encoding resistance to the major antibiotic drug groups. Genes encoding resistance to colistin and the carbapenems were not detected. Genomic surveillance of E. coli in wastewater could be used to monitor new and circulating lineages and resistance determinants of public-health importance.
-
-
-
Introducing BAIT (Biofilm Architecture Inference Tool): a software program to evaluate the architecture of oral multi-species biofilms
More LessBiofilm model systems are used to study biofilm growth and predict the effects of anti-biofilm interventions within the human oral cavity. Many in vitro biofilm model systems use a confocal laser scanning microscope (CLSM) in conjunction with image analysis tools to study biofilms. The aim of this study was to evaluate an in-house developed image analysis software program that we call BAIT (Biofilm Architecture Inference Tool) to quantify the architecture of oral multi-species biofilms following anti-biofilm interventions using a microfluidic biofilm system. Differences in architecture were compared between untreated biofilms and those treated with water (negative control), sodium gluconate (‘placebo’) or stannous fluoride (SnF2). The microfluidic system was inoculated with pooled human saliva and biofilms were developed over 22 h in filter-sterilized 25 % pooled human saliva. During this period, biofilms were treated with water, sodium gluconate, or SnF2 (1000, 3439 or 10 000 p.p.m. Sn2+) 8 and 18 h post-inoculation. After 22 h of growth, biofilms were stained with LIVE/DEAD stain, and imaged by CLSM. BAIT was used to calculate biofilm biovolume, total number of objects, surface area, fluffiness, connectivity, convex hull porosity and viability. Image analysis showed oral biofilm architecture was significantly altered by 3439 and 10 000 p.p.m. Sn2+ treatment regimens, resulting in decreased biovolume, surface area, number of objects and connectivity, while fluffiness increased (P<0.01). In conclusion, BAIT was shown to be able to measure the changes in biofilm architecture and detects possible antimicrobial and anti-biofilm effects of candidate agents.
-
-
-
Synergistic potential of Juniperus communis and Helichrysum italicum essential oils against nontuberculous mycobacteria
More LessObjective. The present study evaluated the possible synergistic antimycobacterial interactions of Juniperus communis and Helichrysum italicum essential oils (EO).
Methods. Antimycobacterial potential was tested against Mycobacterium avium and Mycobacterium intracellulare using broth and water dilution method and checkerboard synergy method. Antiadhesion and antibiofilm effect of EOs was evaluated on biotic (HeLa cells) and abiotic surface (polystyrene). To evaluate the possible mechanisms of action, cellular leakage of proteins and DNA was tested and structural changes were visualized with a transmission electron microscope.
Results. MIC, minimum bactericidal concentration (MBC) and minimal effective concentration (MEC) were 1.6 mg ml−1 for J. communis EO and 3.2 mg ml−1 for H. italicum EO against both mycobacteria. All combinations of EOs in checkerboard synergy method produced fractional inhibitory concentration index values ranging from 0.501 to 1.5, corresponding to synergistic, additive or indifferent effects. Mycobacterium avium showed a greater tendency to create biofilm but these EOs at subinhibitory concentrations (sMIC) effectively blocked the adhesion and the establishment of biofilm. The exposure of both mycobacteria to MICs and sMICs lead to significant morphological changes: acquired a swollen form, ghost-like cell, disorganized cytoplasm detached from the cell wall. OD value of supernatant for both mycobacteria exposed to EOs have confirmed that there is a leakage of cellular material.
Conclusion. The leakage of the cellular material is noticeably higher in sMIC, which is probably due to cell wall damage. sMIC of both EOs have an additive or synergistic effect, reducing MICs, limiting adhesion and preventing the formation of biofilms.
-
-
-
The potential of fosfomycin for multi-drug resistant sepsis: an analysis of in vitro activity against invasive paediatric Gram-negative bacteria
More LessP urpose . Antimicrobial resistance (AMR) is of increasing global concern, threatening to undermine recent progress in reducing child and neonatal mortality. Repurposing older antimicrobials is a prominent strategy to combat multidrug-resistant sepsis. A potential agent is fosfomycin, however, there is scarce data regarding its in vitro activity and pharmacokinetics in the paediatric population.
M ethodology . We analysed a contemporary, systematically collected archive of community-acquired (CA) and hospital-acquired (HA) paediatric Gram-negative bacteraemia isolates for their susceptibility to fosfomcyin. MICs were determined using agar serial dilution methods and validated by disk diffusion testing where breakpoints are available. Disk diffusion antimicrobial susceptibility testing was also conducted for current empirical therapies (ampicillin, gentamicin, ceftriaxone) and amikacin (proposed in the literature as a new combination empirical therapeutic option).
R esults . Fosfomycin was highly active against invasive Gram-negative isolates, including 90 % (202/224) of Enterobacteriaceae and 96 % (22/23) of Pseudomonas spp. Fosfomycin showed high sensitivity against both CA isolates (94 %, 142/151) and HA isolates (81 %, 78/96; P =0.0015). CA isolates were significantly more likely to be susceptible to fosfomycin than the current first-line empirical therapy (96 % vs 59 %, P <0.0001). Extended spectrum β-lactamases (ESBL) production was detected in 34 % (85/247) of isolates with no significant difference in fosfomycin susceptibility between ESBL-positive or -negative isolates [73/85 (86 %) vs 147/162 (91 %) respectively, P =0.245]. All isolates were susceptible to a fosfomycin-amikacin combination.
C onclusion . Gram-negative paediatric bacteraemia isolates are highly susceptible to fosfomycin, which could be combined with aminoglycosides as a new, carbapenem-sparing regimen to achieve excellent coverage to treat antimicrobial-resistant neonatal and paediatric sepsis.
-
-
-
Use of whole genome sequencing in surveillance for antimicrobial-resistant Shigella sonnei infections acquired from domestic and international sources
More LessShigella species are a major cause of gastroenteritis worldwide, and Shigella sonnei is the most common species isolated within the United States. Previous surveillance work in Pennsylvania documented increased antimicrobial resistance (AMR) in S. sonnei associated with reported illnesses. The present study examined a subset of these isolates by whole genome sequencing (WGS) to determine the relationship between domestic and international isolates, to identify genes that may be useful for identifying specific Global Lineages of S. sonnei and to test the accuracy of WGS for predicting AMR phenotype. A collection of 22 antimicrobial-resistant isolates from patients infected within the United States or while travelling internationally between 2009 and 2014 was chosen for WGS. Phylogenetic analysis revealed both international and domestic isolates were one of two previously defined Global Lineages of S. sonnei , designated Lineage II and Lineage III. Twelve of 17 alleles tested distinguish these two lineages. Lastly, genome analysis was used to identify AMR determinants. Genotypic analysis was concordant with phenotypic resistance for six of eight antibiotic classes. For aminoglycosides and trimethoprim, resistance genes were identified in two and three phenotypically sensitive isolates, respectively. This article contains data hosted by Microreact.
-
-
-
Functional characterization of BcrR: a one-component transmembrane signal transduction system for bacitracin resistance
More LessBacitracin is a cell wall targeting antimicrobial with clinical and agricultural applications. With the growing mismatch between antimicrobial resistance and development, it is essential we understand the molecular mechanisms of resistance in order to prioritize and generate new effective antimicrobials. BcrR is a unique membrane-bound one-component system that regulates high-level bacitracin resistance in Enterococcus faecalis . In the presence of bacitracin, BcrR activates transcription of the bcrABD operon conferring resistance through a putative ATP-binding cassette (ABC) transporter (BcrAB). BcrR has three putative functional domains, an N-terminal helix–turn–helix DNA-binding domain, an intermediate oligomerization domain and a C-terminal transmembrane domain. However, the molecular mechanisms of signal transduction remain unknown. Random mutagenesis of bcrR was performed to generate loss- and gain-of-function mutants using transcriptional reporters fused to the target promoter P bcrA . Fifteen unique mutants were isolated across all three proposed functional domains, comprising 14 loss-of-function and one gain-of-function mutant. The gain-of-function variant (G64D) mapped to the putative dimerization domain of BcrR, and functional analyses indicated that the G64D mutant constitutively expresses the P bcrA-luxABCDE reporter. DNA-binding and membrane insertion were not affected in the five mutants chosen for further characterization. Homology modelling revealed putative roles for two key residues (R11 and S33) in BcrR activation. Here we present a new model of BcrR activation and signal transduction, providing valuable insight into the functional characterization of membrane-bound one-component systems and how they can coordinate critical bacterial responses, such as antimicrobial resistance.
-
-
-
Rapid phenotypic evolution in multidrug-resistant Klebsiella pneumoniae hospital outbreak strains
More LessCarbapenem-resistant Klebsiella pneumoniae (CRKP) increasingly cause high-mortality outbreaks in hospital settings globally. Following a patient fatality at a hospital in Beijing due to a bla KPC-2-positive CRKP infection, close monitoring was put in place over the course of 14 months to characterize all bla KPC-2-positive CRKP in circulation in the hospital. Whole genome sequences were generated for 100 isolates from bla KPC-2-positive isolates from infected patients, carriers and the hospital environment. Phylogenetic analyses identified a closely related cluster of 82 sequence type 11 (ST11) isolates circulating in the hospital for at least a year prior to admission of the index patient. The majority of inferred transmissions for these isolates involved patients in intensive care units. Whilst the 82 ST11 isolates collected during the surveillance effort all had closely related chromosomes, we observed extensive diversity in their antimicrobial resistance (AMR) phenotypes. We were able to reconstruct the major genomic changes underpinning this variation in AMR profiles, including multiple gains and losses of entire plasmids and recombination events between plasmids, including transposition of bla KPC-2. We also identified specific cases where variation in plasmid copy number correlated with the level of phenotypic resistance to drugs, suggesting that the number of resistance elements carried by a strain may play a role in determining the level of AMR. Our findings highlight the epidemiological value of whole genome sequencing for investigating multi-drug-resistant hospital infections and illustrate that standard typing schemes cannot capture the extraordinarily fast genome evolution of CRKP isolates.
-
-
-
Very rapid flow cytometric assessment of antimicrobial susceptibility during the apparent lag phase of microbial (re)growth
More Less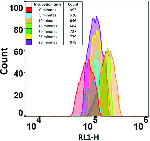 Rapid changes in the number and flow cytometric behaviour of cells of
E. coli
taken from a stationary phase and inoculated into rich medium.
Rapid changes in the number and flow cytometric behaviour of cells of
E. coli
taken from a stationary phase and inoculated into rich medium.Cells of E. coli were grown in LB medium, taken from a stationary phase of 2–4 h, and re-inoculated into fresh media at a concentration (105 ml−1 or lower) characteristic of bacteriuria. Flow cytometry was used to assess how quickly we could detect changes in cell size, number, membrane energization (using a carbocyanine dye) and DNA distribution. It transpired that while the lag phase observable macroscopically via bulk OD measurements could be as long as 4 h, the true lag phase could be less than 15–20 min, and was accompanied by many observable biochemical changes. Antibiotics to which the cells were sensitive affected these changes within 20 min of re-inoculation, providing the possibility of a very rapid antibiotic susceptibility test on a timescale compatible with a visit to a GP clinic. The strategy was applied successfully to genuine potential urinary tract infection (UTI) samples taken from a doctor’s surgery. The methods developed could prove of considerable value in ensuring the correct prescription and thereby lowering the spread of antimicrobial resistance.
-
-
-
Acquired qnrVC1 and bla NDM-1 resistance markers in an international high-risk Pseudomonas aeruginosa ST773 clone
More LessA multidrug-resistant Pseudomonas aeruginosa PS1 isolated from urine clinical sample was investigated in this study. The strain exhibited resistance to piperacillin/tazobactam, ciprofloxacin, imipenem, ceftazidime but it was susceptible to colistin. Analysis of whole-genome sequencing data by ResFinder detected various resistance determinants including qnrVC1 and bla NDM-1. The multiresistant P. aeruginosa isolate belonged to ST773 high-risk clone. The qnrVC1 and bla NDM-1 determinants were incorporated into different integrons. Expression of bla NDM-1 was fourfold and qnrVC1 was twofold increased, compared to that of rpsL housekeeping gene. Mutations in gyrA Thr83Leu and parC Ser87Leu were detected and additionally qnrVC1 expression indicates protective effect of QnrVC1 pentapeptid protein on gyrase and topoisomerase. High-risk P. aeruginosa clones integrate various carbapenemase and other resistance determinants into their genomes that facilitates further dissemination of multiresistance among clinical isolates. We report bla NDM-1 and qnrVC1 genes in P. aeruginosa ST773 international high-risk clone.
-
-
-
Antifungal susceptibilities, biofilms, phospholipase and proteinase activities in the Candida rugosa complex and Candida pararugosa isolated from tertiary teaching hospitals
More LessPurpose. Non-albicans Candida species have emerged as fungal pathogens that cause invasive infections, with many of these species displaying resistance to commonly used antifungal agents. This study was confined to studying the characteristics of clinical isolates of the C. rugosa complex and C. pararugosa species.
Methodology. Seven isolates of the C. rugosa complex and one isolate of C. pararugosa were obtained from two tertiary referral hospitals in Malaysia. Their antifungal susceptibilities, biofilm, proteinase, phospholipase, esterase and haemolysin activities were characterized. Biofilms were quantified using crystal violet (CV) and tetrazolium (XTT) reduction assays at 1.5, 6, 18, 24, 48 and 72 h.
Results/Key findings. The E-test antifungal tests showed that both species have elevated MICs compared to C. albicans and C. tropicalis. The highest biomass was observed in one of the C. rugosa isolates (0.237), followed by C. pararugosa (0.206) at 18 h of incubation. However, the highest bioactivity was observed in the C. rugosa ATCC 10571 strain at 24 h (0.075), followed by C. pararugosa at 48 h (0.048) and the same C. rugosa strain at 24 h (0.046), with P<0.05. All isolates exhibited high proteinase activity (+++) whereas six isolates showed very strong esterase activity (++++). All the isolates were alpha haemolytic producers. None of the isolates exhibited phospholipase activity.
Conclusion. Elevated MICs were shown for the C. rugosa complex and C. pararugosa for commonly used antifungal drugs. Further studies to identify virulence genes involved in the pathogenesis and genes that confer reduced drug susceptibility in these species are proposed.
-
-
-
Australian porcine clonal complex 10 (CC10) Escherichia coli belong to multiple sublineages of a highly diverse global CC10 phylogeny
More LessWe recently identified clonal complex 10 (CC10) Escherichia coli as the predominant clonal group in two populations of healthy Australian food-production pigs. CC10 are highly successful, colonizing humans, food-production animals, fresh produce and environmental niches. Furthermore, E. coli within CC10 are frequently drug resistant and increasingly reported as human and animal extra-intestinal pathogens. In order to develop a high-resolution global phylogeny and determine the repertoire of antimicrobial-resistance genes, virulence-associated genes and plasmid types within this clonal group, we downloaded 228 publicly available CC10 short-read genome sequences for comparison with 20 porcine CC10 we have previously described. Core genome single nucleotide polymorphism phylogeny revealed a highly diverse global phylogeny consisting of multiple lineages that did not cluster by geography or source of the isolates. Australian porcine strains belonged to several of these divergent lineages, indicative that CC10 is present in these animals due to multiple colonization events. Differences in resistance gene and plasmid carriage between porcine strains and the global collection highlighted the role of lateral gene transfer in the evolution of CC10 strains. Virulence profiles typical of extra-intestinal pathogenic E. coli were present in both Australian porcine strains and the broader collection. As both the core phylogeny and accessory gene characteristics appeared unrelated to the geography or source of the isolates, it is likely that the global expansion of CC10 is not a recent event and may be associated with faecal carriage in humans.
-
-
-
Genomic epidemiology of severe community-onset Acinetobacter baumannii infection
More LessAcinetobacter baumannii causes severe, fulminant, community-acquired pneumonia (CAP) in tropical and subtropical regions. We compared the population structure, virulence and antimicrobial resistance determinants of northern Australian community-onset A. baumannii strains with local and global strains. We performed whole-genome sequencing on 55 clinical and five throat colonization A. baumannii isolates collected in northern Australia between 1994 and 2016. Clinical isolates included CAP (n=41), healthcare-associated pneumonia (n=7) and nosocomial bloodstream (n=7) isolates. We also included 93 publicly available international A. baumannii genome sequences in the analyses. Patients with A. baumannii CAP were almost all critically unwell; 82 % required intensive care unit admission and 18 % died during their inpatient stay. Whole-genome phylogenetic analysis demonstrated that community-onset strains were not phylogenetically distinct from nosocomial strains. Some non-multidrug-resistant local strains were closely related to multidrug-resistant strains from geographically distant locations. Pasteur sequence type (ST)10 was the dominant ST and accounted for 31/60 (52 %) northern Australian strains; the remainder belonged to a diverse range of STs. The most recent common ancestor for ST10 was estimated to have occurred in 1738 (95 % highest posterior density, 1626–1826), with evidence of multiple introduction events between Australia and Southeast Asia between then and the present day. Virulence genes associated with biofilm formation and the type 6 secretion system (T6SS) were absent in many strains, and were not associated with in-hospital mortality. All strains were susceptible to gentamicin and meropenem; none carried an AbaR resistance island. Our results suggest that international dissemination of A. baumannii is occurring in the community on a contemporary timescale. Genes associated with biofilm formation and the T6SS may not be required for survival in community niches. The relative contributions of host and bacterial factors to the clinical severity of community-onset A. baumannii infection require further investigation.
-
-
-
Genotypic and phenotypic variations in methicillin-resistant Staphylococcus aureus isolates from outpatient, inpatient and nursing homes
More LessPurpose. The epidemiological shift in MRSA distribution from healthcare-related facilities to the general population is distressing and requires continuous monitoring to manage and control the rate of incidences.
Method. The retrospective relationship between genetic and phenotypic variability of methicillin-resistant Staphylococcus aureus (MRSA) isolates was determined in respect to the specimen source, patient location, sex and age. A total of 521 MRSA isolates were classified based on SCCmec, mec, agr, pvl and spa genetic markers using three different multiplex PCRs.
Results. Based on the genetic variability, the isolates were divided into 97 profiles, of which 59% belonged to only two profiles (P17 and P33). P17 was the predominate profile, harbouring SCCmecIVa, ccr2, mecB, agr1, spa413 and pvl markers. P17 was more prevalent among the younger population (average 33.9 years) from outpatient (77%) locations and wound (88%) sources. The second largest profile was P33, harbouring SCCmecII, ccr2+ccr3, mecA, agr2, spa413 and no PVL. P33 was more prevalent in the older population (average 70.7 years) and more common in females (62%) than males (38%). With respect to antibiotic resistance, P33 exhibited a high rate of resistance to penicillins, cephalosporins, fluoroquinolones and macrolides, and P17 had a lower resistance to fluoroquinolones.
Conclusion. This report contributes to the existing understanding of evolutionary epidemiology of antibiotic resistance in MRSA. The diversity of MRSA isolates and unique environmental preferences for each profile highlights the importance of epidemiological knowledge of MRSA distribution to determine the best treatment for patients in both community and hospital settings.
-
-
-
Oxidative stress response plays a role in antibiotic tolerance of Streptococcus mutans biofilms
More LessKnowledge about biofilm-associated antibiotic tolerance mechanisms is warranted in order to develop effective treatments against biofilm infections. We performed a screen of a Streptococcus mutans transposon mutant library for mutants with reduced biofilm-associated antimicrobial tolerance, and found that the spxA1 gene plays a role in tolerance towards gentamicin and other antibiotics such as vancomycin and linezolid. SpxA1 is a regulator of genes involved in the oxidative stress response in S. mutans . The oxidative stress response genes gor and ahpC were found to be up-regulated upon antibiotic treatment of S. mutans wild-type biofilms, but not spxA1 mutant biofilms. The gor gene product catalyses the formation of glutathione which functions as an important antioxidant during oxidative stress, and accordingly biofilm-associated antibiotic tolerance of the spxA1 mutant could be restored by exogenous addition of glutathione. Our results indicate that the oxidative stress response plays a role in biofilm-associated antibiotic tolerance of S. mutans , and add to the on-going debate on the role of reactive oxygen species in antibiotic mediated killing of bacteria.
-
-
-
Prevalence and genetic characterization of cephalosporin-resistant Enterobacteriaceae among dogs and cats in an animal shelter
More LessPurpose . The prevalence of antimicrobial-resistant bacteria, especially cephalosporin-resistant Enterobacteriaceae , is a major concern for human and animal health. We investigated the prevalence of cephalosporin-resistant Enterobacteriaceae among sheltered dogs and cats with various backgrounds.
Method . Faecal samples or rectal swabs were collected from 151 dogs and 182 cats, and screened for the presence of antimicrobial-resistant bacteria. Isolates were characterized phenotypically and genotypically by pulsed-field gel electrophoresis, multi-locus sequence typing and phylogenetic grouping. The animal attributes related to bacterial carriage were statistically analysed.
Results . Cephalosporin-resistant Enterobacteriaceae was detected in 22 dogs (14.6%) and 20 cats (11.0%): 21 were extended-spectrum β-lactamase (ESBL)-producing, 20 were AmpC-producing, and 1 was both ESBL- and AmpC-producing. Their β-lactamase genes were varied and associated with humans, animals or other origins. The genes CTX-M-14 (n=9) and CMY-2 (n=9) were dominant, but CTX-M-1, CTX-M-2, CTX-M-8, CTX-M-15, CTX-M-24, CTX-M-27, CTX-M-55 and DHA-1 genes were also detected. Genotyping of isolates revealed that β-lactamase-producing Enterobacteriaceae had high genetic diversity. Relationships between animals harbouring cephalosporin-resistant Enterobacteriaceae and individual attributes, such as sex and nutrition type, were detected, but there was no correlation between history of human association and the presence of the bacterium in either dogs or cats.
Conclusion . We found several types of cephalosporin-resistant Enterobacteriaceae distributed among companion animals with a range of individual attributes and histories in Osaka, Japan. Companion animals may play a bridging role in the circulation of antimicrobial-resistant bacteria from humans and from other origins.
-
-
-
Retrospective whole-genome sequencing analysis distinguished PFGE and drug-resistance-matched retail meat and clinical Salmonella isolates
More LessNon-typhoidal Salmonella is a leading cause of outbreak and sporadic-associated foodborne illnesses in the United States. These infections have been associated with a range of foods, including retail meats. Traditionally, pulsed-field gel electrophoresis (PFGE) and antibiotic susceptibility testing (AST) have been used to facilitate public health investigations of Salmonella infections. However, whole-genome sequencing (WGS) has emerged as an alternative tool that can be routinely implemented. To assess its potential in enhancing integrated surveillance in Pennsylvania, USA, WGS was used to directly compare the genetic characteristics of 7 retail meat and 43 clinical historic Salmonella isolates, subdivided into 3 subsets based on PFGE and AST results, to retrospectively resolve their genetic relatedness and identify antimicrobial resistance (AMR) determinants. Single nucleotide polymorphism (SNP) analyses revealed that the retail meat isolates within S. Heidelberg, S. Typhimurium var. O5- subset 1 and S. Typhimurium var. O5- subset 2 were separated from each primary PFGE pattern-matched clinical isolate by 6–12, 41–96 and 21–81 SNPs, respectively. Fifteen resistance genes were identified across all isolates, including fosA7, a gene only recently found in a limited number of Salmonella and a ≥95 % phenotype to genotype correlation was observed for all tested antimicrobials. Moreover, AMR was primarily plasmid-mediated in S. Heidelberg and S. Typhimurium var. O5- subset 2, whereas AMR was chromosomally carried in S. Typhimurium var. O5- subset 1. Similar plasmids were identified in both the retail meat and clinical isolates. Collectively, these data highlight the utility of WGS in retrospective analyses and enhancing integrated surveillance for Salmonella from multiple sources.
-
-
-
Synergistic activity of polymyxin B combined with vancomycin against carbapenem-resistant and polymyxin-resistant Acinetobacter baumannii: first in vitro study
More LessPurpose. The effect of a combination of polymyxin B (PMB) and vancomycin (VAN) was assessed against six Acinetobacter baumannii clinical isolates belonging to six different clusters (three PMB-susceptible and three PMB-resistant).
Methodology. The synergistic effect of the PMB–VAN combination was determined with the checkerboard, time-kill, disk-diffusion and M.I.C.Evaluator assays. PMB-resistance was investigated with mcr-1 gene amplification and a mutant frequency assay.
Results. In the checkerboard assay, all PMB-resistant isolates showed a synergistic effect. The time-kill assay demonstrated that the PMB–VAN combination had a bactericidal effect at 24 h against isolates with a high mutant rate for PMB, suggesting that this combination may block the hypermutation of some isolates. No antagonism was detected. All PMB-resistant isolates also showed synergism in the disk-diffusion test, and a significant decrease in VAN MICs in the M.I.C.Evaluator assay.
Conclusion. Our findings indicate that the PMB–VAN combination has a synergistic effect on A. baumannii , especially against PMB-resistant isolates.
-
-
-
The clinical significance of carbapenem-resistant Klebsiella pneumoniae rectal colonization in critically ill patients: from colonization to bloodstream infection
More LessPurpose . To highlight the clinical significance of carbapenem-resistant Klebsiella pneumoniae (CRKP) rectal colonization by examining the risk factors for CRKP rectal colonization and subsequent bloodstream infection (BSI) in critically ill patients.
Methodology . Prospective study of CRKP rectal colonization in an intensive care unit (ICU) during a 39-month period. CRKP strains isolated from both the blood cultures and corresponding rectal specimens (n=96) of patients were screened by PCR for the presence of antibiotic resistance-associated genes. Molecular analyses were conducted to investigate the clonal relatedness of CRKP strains from the rectal and blood specimens.
Results . Among the 498 patients, 226 were rectally colonized by CRKP, 48 of whom developed a CRKP BSI. The median time from hospital admission to the detection of CRKP rectal colonization was 8 days, while the median time from colonization to BSI was 4 days. The duration of ICU stay, patient/nurse ratio and prior use of antianaerobic antimicrobials were associated with CRKP rectal colonization. No specific factor was associated with BSIs in the colonized patients. The bla KPC-2 gene was detected in all 96 strains, which were all classified as sequence type ST-258. Representative pairs (n=48) of CRKP strains colonizing and infecting the same patient shared the same pulsotype.
Conclusion . Our results indicate that hospitalized patients become infected with their colonizing strains, supporting the strong association between colonization and BSI. Limiting antianaerobic antimicrobial administration, reducing the duration of ICU stay and maintaining a low patient/nurse ratio are possible strategies to restrict rectal CRKP colonization in ICUs.
-
-
-
Utilizing genomic analyses to investigate the first outbreak of van A vancomycin-resistant Enterococcus in Australia with emergence of daptomycin non-susceptibility
More LessIntroduction. The majority of vancomycin-resistant Enterococcus faecium (VREfm) in Australia is of the vanB genotype. An outbreak of vanA VREfm emerged in our haematology/oncology unit between November 2014 and May 2015. The first case of daptomycin non-susceptible E. faecium (DNSEfm) detected was a patient with vanA VREfm bacteraemia who showed clinical failure of daptomycin therapy, prompting microbiologic testing confirming daptomycin non-susceptibility.
Objectives. To describe the patient profiles, antibiotic susceptibility and genetic relatedness of vanA VREfm isolates in the outbreak.
Methods. Chart review of vanA VREfm colonized and infected patients was undertaken to describe the demographics, clinical features and outcomes of therapy. Whole genome sequencing of vanA VREfm isolates involved in the outbreak was conducted to assess clonality.
Results. In total, 29 samples from 24 patients tested positive for vanA VREfm (21 screening swabs and 8 clinical isolates). Five isolates were DNSEfm (four patients colonized, one patient with bacteraemia), with only one patient exposed to daptomycin previously. In silico multi-locus sequence typing of the isolates identified 25/26 as ST203, and 1/26 as ST796. Comparative genomic analysis revealed limited core genome diversity amongst the ST203 isolates, consistent with an outbreak of a single clone of vanA VREfm.
Conclusions. Here we describe an outbreak of vanA VREfm in a haematology/oncology unit. Genomic analysis supports transmission of an ST203 vanA VRE clone within this unit. Daptomycin non-susceptibility in 5/24 patients left linezolid as the only treatment option. Daptomycin susceptibility cannot be assumed in vanA VREfm isolates and confirmatory testing is recommended.
-
-
-
An improved carbapenem inactivation method, CIMTrisII, for carbapenemase production by Gram-negative pathogens
More LessThe modified carbapenem inactivation method (mCIM) is a simple phenotypic screening method for detecting carbapenemase production by Enterobacteriaceae and Pseudomonas aeruginosa . We recently developed another modified carbapenem inactivation method (CIMTris), in which carbapenemase is extracted from bacteria with Tris-HCl buffer, to detect carbapenemase production by Acinetobacter and Pseudomonas species. This study describes an improved carbapenem inactivation method, CIMTrisII, for detecting carbapenemase production by Gram-negative pathogens, including Enterobacteriaceae , Acinetobacter and Pseudomonas species. CIMTrisII was different from CIMTris in the concentration of Meropenem disks (5-µg MEM disks vs. 10-µg MEM disks), the inoculum volume of the bacteria (a 5-µl loopful vs. a 10 µl loopful) and the incubation time (1 vs. 2 h). CIMTrisII showed an overall sensitivity of 99.3 % and an overall specificity of 95.0 % for tested isolates. In comparison, CIMTris showed a sensitivity of 96.1 % and a specificity of 96.3 %, and mCIM showed a sensitivity of 67.1 % and a specificity of 100 %. CIMTrisII is thus deemed useful for detecting carbapenemase production by Gram-negative pathogens.
-
-
-
Antibiotic resistance of Campylobacter jejuni isolates recovered from humans with diarrhoea in Turkey
More LessPurpose. This study was aimed at investigating the occurrence and genetic mechanisms of resistance to ciprofloxacin, tetracycline and erythromycin in clinical isolates of Campylobacter jejuni recovered from human cases of acute gastroenteritis in Turkey.
Methodology. MIC values of each antibiotic were determined with the epsilometer test (E-test). Resistance genes/mutations were first screened by PCR and analysed by subsequent DNA sequencing.
Results. From a total of 152 C. jejuni isolates tested, 113 (74.3%), 38 (25%) and 9 (5.9%) were found to be resistant to ciprofloxacin, tetracycline and erythromycin, respectively. Sequence analysis of ciprofloxacin-resistant isolates showed that all resistant strains (n=113) carried Thr-86-Ile substition in the gyrA gene, which is the most frequently observed mutation in fluoroquinolone-resistant Campylobacter . All of the tetracycline-resistant isolates (n=38) carried the tetO gene. All of the erythromycin-resistant isolates (n=9) harboured the point mutation A2075G in the 23S rRNA gene, which is the most common mutation conferring macrolide resistance in C. jejuni .
Conclusion. The phenotypic susceptibility testing results were found to agree well with those obtained by genetic detection methods for the C. jejuni isolates tested. The findings of this study showed a very high level of resistance to ciprofloxacin and to a lesser extent to tetracycline while resistance to erythromycin remained at a low level. Thus, erythromycin may be considered as the first choice for treatment of Campylobacter infections in this geographical region when indicated.
-
-
-
How actively should we screen for chlamydia and gonorrhoea in MSM and other high-ST-prevalence populations as we enter the era of increasingly untreatable infections? A viewpoint
More LessA number of national and international organizations are advocating more intensive screening for Chlamydia trachomatis and Neisseria gonorrhoeae in high-prevalence populations as a way to reduce the prevalence of these infections. In this article, we review the available evidence and conclude that there is a paucity of evidence to support this approach. We further hypothesize that increasing screening intensity in high-prevalence populations will result in a considerable risk for the emergence of antimicrobial resistance in Neisseria gonorrhoeae and other pathobionts.
-
-
-
Prevalence of macrolide resistance in Treponema pallidum is associated with macrolide consumption
More LessWe hypothesized that the large global variations in the prevalence of macrolide resistance in Treponema pallidum are related to differences in population-level macrolide consumption. The hypothesis was tested by, at a country-level, regressing the peak prevalence of macrolide resistance against the national macrolide consumption in the year prior to this, controlling for the year of the resistance prevalence estimate. A strong association was found between the per capita consumption of macrolides and macrolide resistance (coefficient 0.7, 95 % confidence interval 0.2–0.12, P=0.009).
-
-
-
Evolution of a clade of Acinetobacter baumannii global clone 1, lineage 1 via acquisition of carbapenem- and aminoglycoside-resistance genes and dispersion of ISAba1
More LessResistance to carbapenem and aminoglycoside antibiotics is a critical problem in Acinetobacter baumannii , particularly when genes conferring resistance are acquired by multiply or extensively resistant members of successful globally distributed clonal complexes, such as global clone 1 (GC1) . Here, we investigate the evolution of an expanding clade of lineage 1 of the GC1 complex via repeated acquisition of carbapenem- and aminoglycoside-resistance genes. Lineage 1 arose in the late 1970s and the Tn6168/OCL3 clade arose in the late 1990s from an ancestor that had already acquired resistance to third-generation cephalosporins and fluoroquinolones. Between 2000 and 2002, two distinct subclades have emerged, and they are distinguishable via the presence of an integrated phage genome in subclade 1 and AbaR4 (carrying the oxa23 carbapenem-resistance gene in Tn2006) at a specific chromosomal location in subclade 2. Part or all of the original resistance gene cluster in the chromosomally located AbaR3 has been lost from some isolates, but plasmids carrying alternate resistance genes have been gained. In one group in subclade 2, the chromosomally located AbGRI3, carrying the armA aminoglycoside-resistance gene, has been acquired from a GC2 isolate and incorporated via homologous recombination. ISAba1 entered the common ancestor of this clade as part of the cephalosporin-resistance transposon Tn6168 and has dispersed differently in each subclade. Members of subclade 1 share an ISAba1 in one specific position in the chromosome and in subclade 2 two different ISAba1 locations are shared. Further shared ISAba1 locations distinguish further divisions, potentially providing simple markers for epidemiological studies.
-
-
-
Fosfomycin: mechanisms and the increasing prevalence of resistance
More LessThere are challenges regarding increased global rates of microbial resistance and the emergence of new mechanisms that result in microorganisms becoming resistant to antimicrobial drugs. Fosfomycin is a broad-spectrum bactericidal antibiotic effective against Gram-negative and certain Gram-positive bacteria, such as Staphylococci, that interfere with cell wall synthesis. During the last 40 years, fosfomycin has been evaluated in a wide range of applications and fields. Although numerous studies have been done in this area, there remains limited information regarding the prevalence of resistance. Therefore, in this review, we focus on the available data concerning the mechanisms and increasing resistance regarding fosfomycin.
-
-
-
Multi-step genomic dissection of a suspected intra-hospital Helicobacter cinaedi outbreak
More LessHelicobacter cinaedi is an emerging pathogen causing bacteraemia and cellulitis. Nosocomial transmission of this microbe has been described, but detailed molecular-epidemiological analyses have not been performed. Here, we describe the results of a multi-step genome-wide phylogenetic analysis of a suspected intra-hospital outbreak of H. cinaedi that occurred in a hospital in Japan. The outbreak was recognized by the infectious control team (ICT) of the hospital as a sudden increase in H. cinaedi bacteraemia. ICT defined this outbreak case based on 16S rRNA sequence data and epidemiological information, but were unable to determine the source and route of the infections. We therefore re-investigated this case using whole-genome sequencing (WGS). We first performed a species-wide analysis using publicly available genome sequences to understand the level of genomic diversity of this under-studied species. The clusters identified were then separately analysed using the genome sequence of a representative strain in each cluster as a reference. These analyses provided a high-level phylogenetic resolution of each cluster, identified a confident set of outbreak isolates, and discriminated them from other closely related but distinct clones, which were locally circulating and invaded the hospital during the same period. By considering the epidemiological data, possible strain transmission chains were inferred, which highlighted the role of asymptomatic carriers or environmental contamination. The emergence of a subclone with increased resistance to fluoroquinolones in the outbreak was also recognized. Our results demonstrate the impact of the use of a closely related genome as a reference to maximize the power of WGS.
-
-
-
Transconjugation of erm(X) conferring high-level resistance of clindamycin for Cutibacterium acnes
More LessCutibacterium acnes (C. acnes) can become an exacerbating factor in acne vulgaris. Clindamycin has been most frequently used for the treatment of inflammatory acne vulgaris. We studied clindamycin susceptibility and resistance determinants of C. acnes isolated from acne patients in Japan. The isolation rate of clindamycin-resistant C. acnes had significantly increased from 20.3 % in 2009–2010 to 44.1 % in 2016–2017. Strains carrying erm(X), which confers high-level resistance to clindamycin, had significantly increased from 1.4 to 11.8 %. Sequence analysis of the resistance determinant showed that erm(X) was coded on transposon Tn5432. A transconjugation experiment showed that erm(X) can be transferred between C. acnes strains with high frequency and the transconjugants harboured transposon Tn5432 encoding erm(X). Our data show the transconjugation of erm(X) in C. acnes and strongly suggest that the transmission of erm(X) between C. acnes contributes to the increase and spread of clindamycin-resistant C. acnes strains in acne patients.
-
-
-
A mutation in anti-sigma factor MAB_3542c may be responsible for tigecycline resistance in Mycobacterium abscessus
More LessIn this study, we characterized 7C, a spontaneous mutant selected from tigecycline-susceptible Mycobacterium abscessus ATCC 19977. Whole-genome sequencing (WGS) was used to identify possible resistance determinants in this mutant. Compared to the wild-type, 7C demonstrated resistance to tigecycline as well as cross-resistance to imipenem, and had a slightly retarded growth rate. WGS and subsequent biological verifications showed that these phenotypes were caused by a point mutation in MAB_3542c, which encodes an RshA-like protein. In Mycobacterium tuberculosis, RshA is an anti-sigma factor that negatively regulates the heat/oxidative stress response mechanisms. The MAB_3542c mutation may represent a novel determinant of tigecycline resistance. We hypothesize that this mutation may dysregulate the stress-response pathways which have been shown to be linked to antibiotic resistance in previous studies.
-
-
-
Automated detection of bacterial growth on 96-well plates for high-throughput drug susceptibility testing of Mycobacterium tuberculosis
More LessM. tuberculosis grows slowly and is challenging to work with experimentally compared with many other bacteria. Although microtitre plates have the potential to enable high-throughput phenotypic testing of M. tuberculosis, they can be difficult to read and interpret. Here we present a software package, the Automated Mycobacterial Growth Detection Algorithm (AMyGDA), that measures how much M. tuberculosis is growing in each well of a 96-well microtitre plate. The plate used here has serial dilutions of 14 anti-tuberculosis drugs, thereby permitting the MICs to be elucidated. The three participating laboratories each inoculated 38 96-well plates with 15 known M. tuberculosis strains (including the standard H37Rv reference strain) and, after 2 weeks' incubation, measured the MICs for all 14 drugs on each plate and took a photograph. By analysing the images, we demonstrate that AMyGDA is reproducible, and that the MICs measured are comparable to those measured by a laboratory scientist. The AMyGDA software will be used by the Comprehensive Resistance Prediction for Tuberculosis: an International Consortium (CRyPTIC) to measure the drug susceptibility profile of a large number (>30000) of samples of M. tuberculosis from patients over the next few years.
-
-
-
Characterization of a colistin-resistant Salmonella enterica 4,[5],12:i:- harbouring mcr-3.2 on a variant IncHI-2 plasmid identified in Canada
More LessWe have identified a Salmonella enterica serotype 4,[5],12:i:- containing a mcr-3.2 in a patient who travelled to Thailand 1 month prior to the identification of it in Canada. The isolate was multidrug resistant, but remained susceptible to the carbapenems, amikacin and piperacillin/tazobactam. The mcr-3.2 was carried on a 261 Kb variant of the IncHI2 pWJ1. This report provides further evidence of the emergence of a ST34 colistin-resistant clone.
-
-
-
Genome analysis provides insights into the epidemiology of infection with Flavobacterium psychrophilum among farmed salmonid fish in Sweden
More LessThe pathogen Flavobacterium psychrophilum is a major problem for the expanding salmonid fish farming industry in Sweden as well as worldwide. A better understanding of the phylogeography and infection routes of F. psychrophilum outbreaks could help to improve aquaculture profitability and the welfare of farmed fish while reducing the need for antibiotics. In the present study, high-throughput genome sequencing was applied to a collection of F. psychrophilum isolates (n=38) from outbreaks on fish farms in different regions of Sweden between 1988 and 2016. Antibiotic susceptibility tests were applied to a subset of the isolates and the results correlated to the presence of genetic resistance markers. We show that F. psychrophilum clones are not regionally biased and that new clones with a higher degree of antibiotic resistance have emerged nationwide during the study period. This supports previous theories of the importance of live fish and egg trade as a route of infection. Continuous monitoring of recovered isolates by high-throughput sequencing techniques in the future could facilitate tracing of clones within and between countries, as well as the detection of emergent virulent or antibiotic-resistant clones. This article contains data hosted by Microreact.
-
-
-
Mechanisms of carbapenem resistance in Acinetobacter pittii and Acinetobacter nosocomialis isolates from Thailand
More LessPurpose. The emergence of carbapenem resistance in non-baumannii Acinetobacter has increased in clinical settings worldwide. We investigated the prevalence and mechanisms of carbapenem resistance in A. pittii and A. nosocomialis Thai isolates.
Methodology. Acinetobacter calcoaceticus–Acinetobacter baumannii (Acb) complex isolates were identified by gyrB mulitplex PCR. Carbapenem susceptibilities were studied by the agar dilution method and carbapenemase genes were detected by multiplex PCR. Reductions of the outer membrane proteins (OMPs) were evaluated by SDS-PAGE. Overexpressions of efflux pumps were detected by using efflux pump inhibitors and RT-PCR.
Results. Of the 346 Acb isolates, 22 and 19 were A. pittii and A. nosocomialis, respectively. The carbapenem resistance rates were 22.7 % in A. pittii and 26.3 % in A. nosocomialis. Three carbapenem-resistant A. pittii carried bla OXA-23. One carbapenem-resistant A. pittii harboured bla OXA-58, while another isolate co-harboured bla OXA-58 and bla IMP-14a. bla OXA-58 was also found in three carbapenem-susceptible A. pittii. Five carbapenem-resistant A. nosocomialis carried bla OXA-23. Eighteen A. pittii isolates carried bla OXA-213-like. Reduced OMPs were found in carbapenem-resistant and -susceptible A. pittii carrying bla OXA-58, but were not detected in carbapenem-resistant A. nosocomialis isolates. Overexpression of adeE was found in carbapenem-resistant A. pittii. No efflux pump genes were present in carbapenem-resistant A. nosocomialis.
Conclusion. The major mechanisms of carbapenem resistance in A. pittii and A. nosocomialis were the production of OXA-23 and OXA-58. Overexpression of adeE played a role in carbapenem resistance in A. pittii. Since bla OXA-58 was found in carbapenem-susceptible A. pittii, using carbapenems in the treatment of A. pittii infection should be considered.
-
-
-
Rapid detection of mcr-1 by recombinase polymerase amplification
More LessPurpose. The plasmid-mediated mcr-1 gene conferring colistin resistance has a strong ability to spread. The objective of this study was to establish a rapid and sensitive recombinase polymerase amplification (RPA) method for plasmid-mediated polymyxin-resistant gene mcr-1 detection.
Methods. Using the reported sequence of the mcr-1 gene, we designed specific primers and probes for RPA. Twenty mcr-1-positive strains carrying IncI2/IncHI2/IncX4/IncP plasmids were screened by RPA in this study. The performance of this new assay was compared to that of PCR, TaqMan probe real-time PCR and SYBR Green-based real-time PCR.
Results. Twenty mcr-1-positive samples and three negative samples were tested by RPA and the positive detection rate for the mcr-1-positive samples was 100 %. The detection limit of RPA was approximately 100 fg. Compared with real-time PCR, the RPA assay was more effective due to shorter reaction times, simpler instruments and higher sensitivity, while it had the same high specificity as real-time PCR.
Conclusion. RPA detection based on the mcr-1 gene was successfully applied in our study. The plasmid-mediated mcr-1 gene conferring colistin drug resistance has a strong ability to spread, suggesting the need to further strengthen the detection of this resistance gene in surveillance. Therefore, we require more sensitive detection methods than have previously been available and the RPA assay established in this study meets these detection needs.
-
-
-
TETyper: a bioinformatic pipeline for classifying variation and genetic contexts of transposable elements from short-read whole-genome sequencing data
More LessMuch of the worldwide dissemination of antibiotic resistance has been driven by resistance gene associations with mobile genetic elements (MGEs), such as plasmids and transposons. Although increasing, our understanding of resistance spread remains relatively limited, as methods for tracking mobile resistance genes through multiple species, strains and plasmids are lacking. We have developed a bioinformatic pipeline for tracking variation within, and mobility of, specific transposable elements (TEs), such as transposons carrying antibiotic-resistance genes. TETyper takes short-read whole-genome sequencing data as input and identifies single-nucleotide mutations and deletions within the TE of interest, to enable tracking of specific sequence variants, as well as the surrounding genetic context(s), to enable identification of transposition events. A major advantage of TETyper over previous methods is that it does not require a genome reference. To investigate global dissemination of Klebsiella pneumoniae carbapenemase (KPC) and its associated transposon Tn4401, we applied TETyper to a collection of over 3000 publicly available Illumina datasets containing bla KPC. This revealed surprising diversity, with over 200 distinct flanking genetic contexts for Tn4401, indicating high levels of transposition. Integration of sample metadata revealed insights into associations between geographic locations, host species, Tn4401 sequence variants and flanking genetic contexts. To demonstrate the ability of TETyper to cope with high-copy-number TEs and to track specific short-term evolutionary changes, we also applied it to the insertion sequence IS26 within a defined K. pneumoniae outbreak. TETyper is implemented in python and is freely available at https://github.com/aesheppard/TETyper.
-
-
-
A carbapenem-resistant clinical isolate of Aeromonas hydrophila in Japan harbouring an acquired gene encoding GES-24 β-lactamase
More LessSeveral species of Aeromonas produce the enzyme CphA metallo-β-lactamase. This study describes an isolate of Aeromonas hydrophila harbouring an acquired gene encoding the carbapenemase GES-24. This isolate was obtained from an inpatient in Okinawa, Japan, with no apparent record of travelling overseas. The minimum inhibitory concentrations of carbapenems against this isolate were 8 µg ml−1 for imipenem and 16 µg ml−1 for meropenem. Recombinant GES-24 hydrolyzed all of the tested β-lactams, including imipenem and meropenem. The genomic environment surrounding bla GES-24 was intI1-bla GES-24 -aac(6′)-IIc-qacEdelta1-sulI-orfX-tetR-tetE. This is the first report of A. hydrophila producing a GES-type carbapenemase.
-
-
-
A genomic view of experimental intraspecies and interspecies transformation of a rifampicin-resistance allele into Neisseria meningitidis
More LessThe spread of antibiotic resistance within and between different bacterial populations is a major health problem on a global scale. The identification of genetic transformation in genomic data from Neisseria meningitidis, the meningococcus (Mc), and other bacteria is problematic, since similar or even identical alleles may be involved. A particular challenge in naturally transformable bacteria generally is to distinguish between common ancestry and true recombined sites in sampled genome sequences. Furthermore, the identification of recombination following experimental transformation of homologous alleles requires identifiable differences between donor and recipient, which in itself influences the propensity for homologous recombination (HR). This study identifies the distribution of HR events following intraspecies and interspecies Mc transformations of rpoB alleles encoding rifampicin resistance by whole-genome DNA sequencing and single nucleotide variant analysis. The HR events analysed were confined to the genomic region surrounding the single nucleotide genetic marker used for selection. An exponential length distribution of these recombined events was found, ranging from a few nucleotides to about 72 kb stretches. The lengths of imported sequences were on average found to be longer following experimental transformation of the recipient with genomic DNA from an intraspecies versus an interspecies donor (P<0.001). The recombination events were generally observed to be mosaic, with donor sequences interspersed with recipient sequence. Here, we present four models to explain these observations, by fragmentation of the transformed DNA, by interruptions of the recombination mechanism, by secondary recombination of endogenous self-DNA, or by repair/replication mechanisms.
-
-
-
Disrupting folate metabolism reduces the capacity of bacteria in exponential growth to develop persisters to antibiotics
More LessBacteria can survive high doses of antibiotics through stochastic phenotypic diversification. We present initial evidence that folate metabolism could be involved with the formation of persisters. The aberrant expression of the folate enzyme gene fau seems to reduce the incidence of persisters to antibiotics. Folate-impaired bacteria had a lower generation rate for persisters to the antibiotics ampicillin and ofloxacin. Persister bacteria were detectable from the outset of the exponential growth phase in the complex media. Gene expression analyses tentatively showed distinctive profiles in exponential growth at times when bacteria persisters were observed. Levels of persisters were assessed in bacteria with altered, genetically and pharmacologically, folate metabolism. This work shows that by disrupting folate biosynthesis and usage, bacterial tolerance to antibiotics seems to be diminished. Based on these findings there is a possibility that bacteriostatic antibiotics such as anti-folates could have a role to play in clinical settings where the incidence of antibiotic persisters seems to drive recalcitrant infections.
-
-
-
Prevalence and antimicrobial susceptibilities of Acinetobacter baumannii and non-baumannii Acinetobacters from Terengganu, Malaysia and their carriage of carbapenemase genes
More LessA total of 153 non-repeat Acinetobacter spp. clinical isolates obtained in 2015 from Hospital Sultanah Nur Zahirah (HSNZ) in Terengganu, Malaysia, were characterized. Identification of the isolates at species level was performed by ribosomal DNA restriction analysis (ARDRA) followed by sequencing of the rpoB gene. The majority of the isolates (n=128; 83.7 %) were A. baumannii while the rest were identified as A. nosocomialis (n=16), A. calcoaceticus (n=5), A. soli (n=2), A. berezeniae (n=1) and A. variabilis (n=1). Multidrug resistance (MDR) was most prevalent in A. baumannnii (66.4 %) whereas only one non-baumannii isolate (A. nosocomialis) was MDR. The bla OXA-23 gene was the predominant acquired carbapenemase gene (56.2 %) and was significantly associated (P<0.001) with carbapenem resistance. However, no significant association was found for carbapenem resistance and isolates that contained the ISAba1-bla OXA-51 configuration.
-
-
-
Salmonella infection – prevention and treatment by antibiotics and probiotic yeasts: a review
More LessGlobal Salmonella infection, especially in developing countries, is a health and economic burden. The use of antibiotic drugs in treating the infection is proving less effective due to the alarming rise of antibiotic-resistant strains of Salmonella, the effects of antibiotics on normal gut microflora and antibiotic-associated diarrhoea, all of which bring a growing need for alternative treatments, including the use of probiotic micro-organisms. However, there are issues with probiotics, including their potential to be opportunistic pathogens and antibiotic-resistant carriers, and their antibiotic susceptibility if used as complementary therapy. Clinical trials, animal trials and in vitro investigations into the prophylactic and therapeutic efficacies of probiotics have demonstrated antagonistic properties against Salmonella and other enteropathogenic bacteria. Nonetheless, there is a need for further studies into the potential mechanisms, efficacy and mode of delivery of yeast probiotics in Salmonella infections. This review discusses Salmonella infections and treatment using antibiotics and probiotics.
-
-
-
The resistomes of six carbapenem-resistant pathogens – a critical genotype–phenotype analysis
More LessCarbapenem resistance is a rapidly growing threat to our ability to treat refractory bacterial infections. To understand how carbapenem resistance is mobilized and spread between pathogens, it is important to study the genetic context of the underlying resistance mechanisms. In this study, the resistomes of six clinical carbapenem-resistant isolates of five different species – Acinetobacter baumannii, Escherichia coli, two Klebsiella pneumoniae, Proteus mirabilis and Pseudomonas aeruginosa – were characterized using whole genome sequencing. All Enterobacteriaceae isolates and the A. baumannii isolate had acquired a large number of antimicrobial resistance genes (7–18 different genes per isolate), including the following encoding carbapenemases: bla KPC-2, bla OXA-48, bla OXA-72, bla NDM-1, bla NDM-7 and bla VIM-1. In addition, a novel version of bla SHV was discovered. Four new resistance plasmids were identified and their fully assembled sequences were verified using optical DNA mapping. Most of the resistance genes were co-localized on these and other plasmids, suggesting a risk for co-selection. In contrast, five out of six carbapenemase genes were present on plasmids with no or few other resistance genes. The expected level of resistance – based on acquired resistance determinants – was concordant with measured levels in most cases. There were, however, several important discrepancies for four of the six isolates concerning multiple classes of antibiotics. In conclusion, our results further elucidate the diversity of carbapenemases, their mechanisms of horizontal transfer and possible patterns of co-selection. The study also emphasizes the difficulty of using whole genome sequencing for antimicrobial susceptibility testing of pathogens with complex genotypes.
-
-
-
mlplasmids: a user-friendly tool to predict plasmid- and chromosome-derived sequences for single species
More LessAssembly of bacterial short-read whole-genome sequencing data frequently results in hundreds of contigs for which the origin, plasmid or chromosome, is unclear. Complete genomes resolved by long-read sequencing can be used to generate and label short-read contigs. These were used to train several popular machine learning methods to classify the origin of contigs from Enterococcus faecium, Klebsiella pneumoniae and Escherichia coli using pentamer frequencies. We selected support-vector machine (SVM) models as the best classifier for all three bacterial species (F1-score E. faecium=0.92, F1-score K. pneumoniae=0.90, F1-score E. coli=0.76), which outperformed other existing plasmid prediction tools using a benchmarking set of isolates. We demonstrated the scalability of our models by accurately predicting the plasmidome of a large collection of 1644 E. faecium isolates and illustrate its applicability by predicting the location of antibiotic-resistance genes in all three species. The SVM classifiers are publicly available as an R package and graphical-user interface called ‘mlplasmids’. We anticipate that this tool may significantly facilitate research on the dissemination of plasmids encoding antibiotic resistance and/or contributing to host adaptation.
-
-
-
Changing the paradigm for hospital outbreak detection by leading with genomic surveillance of nosocomial pathogens
More LessThe current paradigm for hospital outbreak detection and investigation is based on methodology first developed over 150 years ago. Daily surveillance to detect patients positive for pathogens of particular importance for nosocomial infection is supported by epidemiological investigation to determine their relationship in time and place, and to identify any other factor that could link them. The antibiotic resistance pattern is commonly used as a surrogate for bacterial relatedness, although this lacks sensitivity and specificity. Typing may be used to define bacterial relatedness, although routine methods lack sufficient discriminatory power to distinguish relatedness beyond the level of bacterial clones. Ultimately, the identification of an outbreak remains a predominately subjective process reliant on the intuition of experienced infection control professionals. Here, we propose a redesign of hospital outbreak detection and investigation in which bacterial species associated with nosocomial transmission and infection undergo routine prospective whole-genome sequencing. Further investigation is based on the probability that isolates are associated with an outbreak, which is based on the degree of genetic relatedness between isolates. Evidence is provided that supports this model based on studies of MRSA (methicillin-resistant Staphylococcus aureus), together with the benefits of a ‘Sequence First’ approach. The feasibility of implementation is discussed, together with residual barriers that need to be overcome prior to implementation.
-
-
-
Evaluation of rapid KPC carbapenemase detection method based on MALDI-TOF VITEK MS spectra analysis
More LessClinical microbiology laboratories in hospital settings need to be able to identify patients who carry carbapenemase-producing bacterial strains quickly in order to contain their spread and initiate proper pharmacological therapy. The aim of this study was to confirm the correlation between KPC production and a characteristic mass spectrometry (MS) peak (11 109 Da±8) to validate the use of matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) MS as a rapid screening tool. With this aim, 176 selected clinical samples that were KPC-producing and 260 control samples that were carbapenem-susceptible or carbapenem-resistant through other resistance mechanisms, or were producing hydrolytic enzymes other than KPC, were analysed. The presence of the 11 109 Da peak in the spectra of 99.4 % (175/176) of the KPC-producing strains compared to the controls, which all lacked the peak, confirmed a strong correlation between KPC production and the presence of the 11 109 Da peak in the MALDI-TOF MS spectrum. The high sensitivity (98.7 %) and specificity (100 %) of the peak searching in the MALDI-TOF MS spectra mean that 11 109 Da peak searching is a suitable screening tool in KPC-endemic regions.
-
-
-
Excess body weight and age associated with the carriage of fluoroquinolone and third-generation cephalosporin resistance genes in commensal Escherichia coli from a cohort of urban Vietnamese children
More LessPurpose. Antimicrobial-resistant bacterial infections in low- and middle-income countries (LMICs) are a well-established global health issue. We aimed to assess the prevalence of and epidemiological factors associated with the carriage of ciprofloxacin- and ceftriaxone-resistant Escherichia coli and associated resistance genes in a cohort of 498 healthy children residing in urban Vietnam.
Methodology. We cultured rectal swabs onto MacConkey agar supplemented with resistant concentrations of ciprofloxacin and ceftriaxone. Additionally, we screened meta-E. coli populations by conventional PCR to detect plasmid-mediated quinolone resistance (PMQR)- and extended-spectrum β-lactamase (ESBL)-encoding genes. We measured the associations between phenotypic/genotypic resistance and demographic characteristics using logistic regression.
Results/Key findings. Ciprofloxacin- and ceftriaxone-resistant E. coli were cultured from the faecal samples of 67.7 % (337/498) and 80.3 % (400/498) of children, respectively. The prevalence of any associated resistance marker in the individual samples was 86.7 % (432/498) for PMQR genes and 90.6 % (451/498) for β-lactamase genes. Overweight children were significantly more likely to carry qnr genes than children with lower weight-for-height z-scores [odds ratios (OR): 1.24; 95 % confidence interval (CI): 10.5–1.48 for each unit increase in weight for height; P=0.01]. Additionally, younger children were significantly more likely to carry ESBL CTX-M genes than older children (OR: 0.97, 95 % CI: 0.94–0.99 for each additional year, P=0.01).
Conclusion. The carriage of genotypic and phenotypic antimicrobial resistance is highly prevalent among E. coli in healthy children in the community in Vietnam. Future investigations on the carriage of antimicrobial resistant organisms in LMICs should focus on the progression of carriage from birth and structure of the microbiome in obesity.
-
-
-
MinION nanopore sequencing identifies the position and structure of bacterial antibiotic resistance determinants in a multidrug-resistant strain of enteroaggregative Escherichia coli
More LessThe aim of this study was to use single-molecule, nanopore sequencing to explore the genomic environment of the resistance determinants in a multidrug-resistant (MDR) strain of enteroaggregative Escherichia coli serotype O51 : H30, sequence type (ST) 38. Sequencing was performed on the MinION Flow cell MIN-106 R9.4. Nanopore raw FAST5 reads were base-called using Albacore v1.2.1, converted to FASTA and FASTQ formats using Poretools v0.6.0, and assembled using Unicycler v0.4.2, combining the long-read sequencing data with short-read data produced by Illumina sequencing. The genome was interrogated against an antimicrobial resistance (AMR) gene reference database using blast. The majority of the 12 AMR determinants identified were clustered together on the chromosome at three separate locations flanked by integrases and/or insertion elements [region 1 –catA, bla OXA-1, aac(6′)-Ib-cr, tetA and bla CTX-M-15; region 2 – dfrA1 and aadA1; region 3 – catA, bla TEM-1, tetA and sul2]. AMR determinants located outside these three regions were a chromosomally encoded bla CMY-16, mutations in gyrA and parC, and two plasmid-encoded AMR determinants, bla OXA-181 and qnrS1 located on the same IncX3 plasmid. Long-read analysis of whole genome sequencing data identified mobile genetic elements on which AMR determinants were located and revealed the combination of different AMR determinants co-located on the same mobile element. These data contribute to a better understanding of the transmission of co-located AMR determinants in MDR E. coli causing gastrointestinal and extra-intestinal infections.
-
-
-
Resistance pattern and distribution of carbapenemase and antiseptic resistance genes among multidrug-resistant Acinetobacter baumannii isolated from intensive care unit patients
More LessPurpose. Nosocomial infections caused by multidrug resistant Acinetobacter baumannii have emerged as a serious problem in healthcare settings worldwide.
Methodology. A total of 100 A. baumannii clinical isolates from immunocompromised patients hospitalized in ICUs in Iran were investigated for antimicrobial susceptibility and the presence of carbapenemase and antiseptic resistance genes.
Results. All isolates were resistant to one or more antibiotics, with the most frequent resistance found against ciprofloxacin and imipenem (100 %) and piperacillin (99 %). The MICs of biocides were determined by the agar dilution method. No apparent resistance to biocides was seen among the 100 A. baumannii isolates. All isolates were effectively inhibited by the user’s defined concentrations of cetrimide, benzalkonium chloride and glutardaldehyde. The intrinsic β-lactamase gene, bla OXA-51-like, was detected in all A. baumannii isolates. Coexistence of bla OXA-51 andbla OXA-23 was encountered in 89 % of isolates. However, genes bla OXA-58, bla SIM and bla IMP were not detected in any isolates. While A. baumannii isolates were sensitive to biocides, they carried qac genes with the qacEΔ1 gene being the most common, at a frequency of 91 %.
Conclusion. Our study revealed the high frequency of multidrug- and carbapenem-resistant isolates of A. baumannii in ICU patients, with a high prevalence of the genes bla OXA-23 and bla OXA-51. However, no apparent biocide resistance was seen in A. baumannii isolates. It appears that appropriate surveillance and control measures are essential to prevent the emergence and transmission of MDR A. baumannii in Iran.
-
-
-
Broad-spectrum antimicrobial activity by Burkholderia cenocepacia TAtl-371, a strain isolated from the tomato rhizosphere
More LessThe Burkholderia cepacia complex (Bcc) comprises a group of 24 species, many of which are opportunistic pathogens of immunocompromised patients and also are widely distributed in agricultural soils. Several Bcc strains synthesize strain-specific antagonistic compounds. In this study, the broad killing activity of B. cenocepacia TAtl-371, a Bcc strain isolated from the tomato rhizosphere, was characterized. This strain exhibits a remarkable antagonism against bacteria, yeast and fungi including other Bcc strains, multidrug-resistant human pathogens and plant pathogens. Genome analysis of strain TAtl-371 revealed several genes involved in the production of antagonistic compounds: siderophores, bacteriocins and hydrolytic enzymes. In pursuit of these activities, we observed growth inhibition of Candida glabrata and Paraburkholderia phenazinium that was dependent on the iron concentration in the medium, suggesting the involvement of siderophores. This strain also produces a previously described lectin-like bacteriocin (LlpA88) and here this was shown to inhibit only Bcc strains but no other bacteria. Moreover, a compound with an m/z 391.2845 with antagonistic activity against Tatumella terrea SHS 2008T was isolated from the TAtl-371 culture supernatant. This strain also contains a phage-tail-like bacteriocin (tailocin) and two chitinases, but the activity of these compounds was not detected. Nevertheless, the previous activities are not responsible for the whole antimicrobial spectrum of TAtl-371 seen on agar plates, suggesting the presence of other compounds yet to be found. In summary, we observed a diversified antimicrobial activity for strain TAtl-371 and believe it supports the biotechnological potential of this Bcc strain as a source of new antimicrobials.
-
-
-
Cinnamaldehyde disrupts biofilm formation and swarming motility of Pseudomonas aeruginosa
More LessBacterial biofilms can cause serious health care complications associated with increased morbidity and mortality. There is an urge to discover and develop new biofilm inhibitors from natural products or by modifying natural compounds or understanding the modes of action of existing compounds. Cinnamaldehyde (CAD), one of the major components of cinnamon oil, has been demonstrated to act as an antimicrobial agent against a number of Gram-negative and Gram-positive pathogens, including Pseudomonas aeruginosa, Helicobacter pylori and Listeria monocytogenes. Despite the mechanism of action of CAD against the model organism P. aeruginosa being undefined, based on its antimicrobial properties, we hypothesized that it may disrupt preformed biofilms of P. aeruginosa. The minimum inhibitory concentration (MIC) of CAD for planktonic P. aeruginosa was determined to be 11.8 mM. Membrane depolarization assays demonstrated disruption of the transmembrane potential of P. aeruginosa. CAD at 5.9 mM (0.5 MIC) disrupted preformed biofilms by 75.6 % and 3 mM CAD (0.25 MIC) reduced the intracellular concentrations of the secondary messenger, bis-(3′–5′)-cyclic dimeric guanosine monophosphate (c-di-GMP), which controls P. aeruginosa biofilm formation. The swarming motility of P. aeruginosa was also reduced by CAD in a concentration-dependent manner. Collectively, these findings show that sub-MICs of CAD can disrupt biofilms and other surface colonization phenotypes through the modulation of intracellular signalling processes.
-
-
-
Evaluation of the FilmArray Blood Culture Identification Panel compared to direct MALDI-TOF MS identification for rapid identification of pathogens
More LessTo improve time to identification of pathogens and detection of resistance genes, we evaluated the BioFire FilmArray Blood Culture Identification Panel (BCID) as compared to: (1) direct MALDI-TOF MS (DM) and (2) standardized culture-based identification (ID) with antibiotic susceptibility testing (AST). BCID gave an accurate identification in 102/112 (91 %) of cases (102/103 for on-panel organisms). DM gave an accurate identification in 91/112 (81 %) of cases, with 13/91 (14 %) requiring repeat testing from the residual pellet. The mean time to an identification result was 2.4 and 2.9 h for BCID and DM, respectively. Standardized ID and AST results were available at a mean time of 26.5 and 33 h, respectively. There were 44 BCID/DM results that had an antimicrobial treatment change made based on rapid identification and resistant gene detection of pathogens. Both BCID and DM are accurate and rapid methods for the identification of new positive blood culture pathogens.
-
-
-
Genetic diversity, mobilisation and spread of the yersiniabactin-encoding mobile element ICEKp in Klebsiella pneumoniae populations
More LessMobile genetic elements (MGEs) that frequently transfer within and between bacterial species play a critical role in bacterial evolution, and often carry key accessory genes that associate with a bacteria’s ability to cause disease. MGEs carrying antimicrobial resistance (AMR) and/or virulence determinants are common in the opportunistic pathogen Klebsiella pneumoniae, which is a leading cause of highly drug-resistant infections in hospitals. Well-characterised virulence determinants in K. pneumoniae include the polyketide synthesis loci ybt and clb (also known as pks), encoding the iron-scavenging siderophore yersiniabactin and genotoxin colibactin, respectively. These loci are located within an MGE called ICEKp, which is the most common virulence-associated MGE of K. pneumoniae, providing a mechanism for these virulence factors to spread within the population. Here we apply population genomics to investigate the prevalence, evolution and mobility of ybt and clb in K. pneumoniae populations through comparative analysis of 2498 whole-genome sequences. The ybt locus was detected in 40 % of K. pneumoniae genomes, particularly amongst those associated with invasive infections. We identified 17 distinct ybt lineages and 3 clb lineages, each associated with one of 14 different structural variants of ICEKp. Comparison with the wider population of the family Enterobacteriaceae revealed occasional ICEKp acquisition by other members. The clb locus was present in 14 % of all K. pneumoniae and 38.4 % of ybt+ genomes. Hundreds of independent ICEKp integration events were detected affecting hundreds of phylogenetically distinct K. pneumoniae lineages, including at least 19 in the globally-disseminated carbapenem-resistant clone CG258. A novel plasmid-encoded form of ybt was also identified, representing a new mechanism for ybt dispersal in K. pneumoniae populations. These data indicate that MGEs carrying ybt and clb circulate freely in the K. pneumoniae population, including among multidrug-resistant strains, and should be considered a target for genomic surveillance along with AMR determinants.
-
-
-
Genomic reorganization by IS26 in a bla NDM-5-bearing FII plasmid of Klebsiella pneumoniae isolated from a patient in Japan
More LessAn NDM-5-producing Klebsiella pneumoniae ST147 strain was isolated from a Japanese patient who had not travelled abroad in at least 5 years. Whole-genome sequencing revealed a genomic rearrangement in an FII plasmid harbouring bla NDM-5 due to the replicative transposition of IS26. A hypothetical structure was proposed for its ancestral plasmid, and comparative genomic analysis of the plasmid suggested the dissemination of structurally similar plasmids harbouring bla NDM-5 in Asian and Middle Eastern countries.
-
-
-
In vitro activity of β-lactams in combination with avibactam against multidrug-resistant Pseudomonas aeruginosa, Stenotrophomonas maltophilia and Achromobacter xylosoxidans isolates from patients with cystic fibrosis
More LessThe in vitro activity of anti-pseudomonal β-lactams in combination with avibactam was evaluated against 54 multidrug-resistant non-fermenting Gram-negative bacilli isolated from cystic fibrosis patients. Avibactam increased and/or restored the antibacterial activities of ceftazidime and aztreonam against Pseudomonas aeruginosa and Stenotrophomonas maltophilia, respectively. No β-lactam–avibactam combination was active against Achromobacter xylosoxidans.
-
-
-
Molecular determinants of Burkholderia pseudomallei BpeEF-OprC efflux pump expression
More LessBurkholderia pseudomallei, the cause of melioidosis, is intrinsically resistant to many antibiotics. Acquired multidrug resistance, including resistance to doxycycline and co-trimoxazole used for melioidosis eradication phase therapy, is mainly attributed to constitutive expression of the BpeEF-OprC efflux pump. Constitutive expression of this pump is caused by mutations affecting two highly similar LysR-type transcriptional regulators (LTTR), BpeT and BpeS, but their interaction with the regulatory region governing BpeEF-OprC expression has not yet been studied. The bpeE-bpeF-oprC genes are distally located in the llpE-bpeE-bpeF-oprC operon. The llpE gene encodes a putative lipase/esterase of unknown function. We show that in a bpeT mutant llpE is constitutively co-transcribed with bpeE-bpeF-oprC. As expected from previous studies with B. cenocepacia, deletion of llpE does not affect antibiotic efflux. Using transcriptional bpeE′-lacZ fusions, we demonstrate that the 188 bp bpeT-llpE intergenic region located between bpeT and the llpE-bpeE-bpeF-oprC operon contains regulatory elements needed for control of bpeT and llpE-bpeE-bpeF-oprC operon expression. By native polyacrylamide gel electrophoresis and electrophoretic mobility shift assays with purified recombinant BpeT and BpeS proteins, we show BpeT and BpeS form oligomers that share a 14 bp binding site overlapping the essential region required for llpE-bpeE-bpeF-oprC expression. The binding site contains the conserved T-N11-A LTTR box motif involved in binding of LysR proteins, which in concert with two other possible LTTR boxes may mediate BpeT and BpeS regulation of BpeEF-OprC expression. These studies form the basis for further investigation of BpeEF-OprC expression and regulation at the molecular level by yet unknown external stimuli.
-
-
-
Multicenter assessment of the rapid Unyvero Blood Culture molecular assay
More LessPurpose. Bloodstream infections remain an important cause of morbidity and mortality. Rapid diagnosis can reduce the time from empiric antimicrobial therapy to targeted therapy and improve patient outcomes.
Methodology. The fully automated Unyvero Blood Culture (BCU) Application (Curetis GmbH) can identify a broad panel of pathogens (36 analytes covering over 50 pathogens) and 16 antibiotic resistance gene markers simultaneously in about 5 h. The assay was evaluated in three clinical laboratories in comparison to routine microbiological procedures.
Results. A total of 207 blood cultures were included in the study, and 90.5 % of the species identified by culture were covered by the Unyvero BCU panel with an overall sensitivity of 96.8 % and specificity of 99.8 %. The time to result was reduced on average by about 34 h. The assay accurately identified 95 % of the species, including 158/164 monomicrobial and 7/9 polymicrobial cultures. The Unyvero BCU Cartridge detected a large number of resistance markers including mecA (n=57), aac(6′)aph(2′′) (n=40), one vanB resistance gene, and six instances of bla CTX-M.
Conclusion. The Unyvero BCU Application provided fast, reliable results, while significantly improving turnaround time in blood culture diagnostics.
-
-
-
PlaScope: a targeted approach to assess the plasmidome from genome assemblies at the species level
More LessPlasmid prediction may be of great interest when studying bacteria of medical importance such as Enterobacteriaceae as well as Staphylococcus aureus or Enterococcus. Indeed, many resistance and virulence genes are located on such replicons with major impact in terms of pathogenicity and spreading capacities. Beyond strain outbreak, plasmid outbreaks have been reported in particular for some extended-spectrum beta-lactamase- or carbapenemase-producing Enterobacteriaceae. Several tools are now available to explore the ‘plasmidome’ from whole-genome sequences with various approaches, but none of them are able to combine high sensitivity and specificity. With this in mind, we developed PlaScope, a targeted approach to recover plasmidic sequences in genome assemblies at the species or genus level. Based on Centrifuge, a metagenomic classifier, and a custom database containing complete sequences of chromosomes and plasmids from various curated databases, PlaScope classifies contigs from an assembly according to their predicted location. Compared to other plasmid classifiers, PlasFlow and cBar, it achieves better recall (0.87), specificity (0.99), precision (0.96) and accuracy (0.98) on a dataset of 70 genomes of Escherichia coli containing plasmids. In a second part, we identified 20 of the 21 chromosomal integrations of the extended-spectrum beta-lactamase coding gene in a clinical dataset of E. coli strains. In addition, we predicted virulence gene and operon locations in agreement with the literature. We also built a database for Klebsiella and correctly assigned the location for the majority of resistance genes from a collection of 12 Klebsiella pneumoniae strains. Similar approaches could also be developed for other well-characterized bacteria.
-
-
-
Pyrosequencing: a rapid and effective sequencing method to diagnose drug-resistant tuberculosis
More LessPurpose. This study was undertaken to evaluate the efficiency of the pyrosequencing (PSQ) assay for the rapid detection of resistance to rifampicin (RIF), fluoroquinolones (FQs) and second-line injectables (SLIs) such as capreomycin (CAP) and kanamycin (KAN) in Mycobacterium tuberculosis (Mtb) clinical isolates.
Methodology. Pyrosequencing is a simple and accurate short read DNA sequencing method for genome analysis. DNA extraction from Mtb clinical isolates was performed using Tris-HCl buffer and chloroform. The rpoB (RIF), gyrA (FQs) and rrs (aminoglycosides) genes were amplified, followed by sequencing using the PyroMark Q24 ID system. The PSQ results were compared with the results from the conventional drug susceptibility testing performed in the laboratory.
Results. The sensitivity of the PSQ assay for the detection of resistance to RIF, FQ, CAP and KAN was 100 %, 100 %, 40 % and 50 %, respectively. The specificity of the PSQ assay was 100 %.
Conclusion. The PSQ assay is a rapid and effective method for detecting drug resistance mutations from Mtb clinical isolates in a short period of time.
-
-
-
The endogenous antiseptic N-chlorotaurine irreversibly inactivates Chlamydia pneumoniae and Chlamydia trachomatis
More LessPurpose. The antimicrobial activity of N-chlorotaurine (NCT), an endogenous long-lived oxidant applied topically, was tested against Chlamydiae in vitro.
Methodology. Elementary bodies of Chlamydia pneumoniae strain CV-6 and Chlamydia trachomatis serovars A and D were incubated in 0.01, 0.1 and 1 % (w/v) NCT solution at pH 7.1 and 37 °C. After different incubation times, aliquots were removed and grown in cell culture. The number of inclusion forming units was quantified by immunofluorescence and real-time qPCR.
Results/Key findings. Chlamydia pneumoniae and Chlamydia trachomatis were inactivated by 1 and 0.1 % NCT within 1 min. Moreover, 0.025–0.1 % NCT significantly reduced the number of intracellularly growing C. pneumoniae within 30 min.
Conclusions. This is the first study demonstrating the antimicrobial activity of NCT against Chlamydiae. Clinical implications of these findings have to be investigated in further trials.
-
-
-
The return of Pfeiffer’s bacillus: Rising incidence of ampicillin resistance in Haemophilus influenzae
More LessHaemophilus influenzae, originally named Pfeiffer’s bacillus after its discoverer Richard Pfeiffer in 1892, was a major risk for global health at the beginning of the 20th century, causing childhood pneumonia and invasive disease as well as otitis media and other upper respiratory tract infections. The implementation of the Hib vaccine, targeting the major capsule type of H. influenzae, almost eradicated the disease in countries that adapted the vaccination scheme. However, a rising number of infections are caused by non-typeable H. influenzae (NTHi), which has no capsule and against which the vaccine therefore provides no protection, as well as other serotypes equally not recognised by the vaccine. The first line of treatment is ampicillin, but there is a steady rise in ampicillin resistance. This is both through acquired as well as intrinsic mechanisms, and is cause for serious concern and the need for more surveillance. There are also increasing reports of new modifications of the intrinsic ampicillin-resistance mechanism leading to resistance against cephalosporins and carbapenems, the last line of well-tolerated drugs, and ampicillin-resistant H. influenzae was included in the recently released priority list of antibiotic-resistant bacteria by the WHO. This review provides an overview of ampicillin resistance prevalence and mechanisms in the context of our current knowledge about population dynamics of H. influenzae.
-
-
-
Time-kill kinetics of cadazolid and comparator antibacterial agents against different ribotypes of Clostridium difficile
More LessPurpose. Clostridium difficile infection (CDI) is an increasing cause of nosocomial diarrhoea worldwide, which has been partly attributed to the emergence of hypervirulent strains including C. difficile BI/NAP1/ribotype 027 and BK/NAP7/ribotype 078. Cadazolid is a new antibiotic currently in late-stage clinical studies for the treatment of CDI. The present study evaluated the in vitro bactericidal effect of cadazolid and comparator antibiotics against four C. difficile strains. The data demonstrate the potent and bactericidal activity of cadazolid against different ribotypes of C. difficile.
Methodology. MICs for test antibiotics were determined in brain– heart infusion-supplemented broth (BHIS) containing 5 g l−1 yeast extract and 0.025 % (w/v) l-cysteine. Time-kill kinetics to investigate the rate of killing of each antibiotic at sub- and supra-MIC concentrations were performed at concentrations of 0.5, 1, 2, 4, 8 or 16× the MIC of cadazolid, vancomycin and fidaxomicin at intervals over a 48 h period.
Results/key findings. Cadazolid-mediated killing of C. difficile was faster and occurred at lower concentrations than observed for vancomycin, while potency and killing was largely comparable to those observed for fidaxomicin. Notably, cadazolid also displayed a potent bactericidal effect against fluoroquinolone-resistant hypervirulent ribotype 027 and 078 strains. C. difficile spore formation was largely inhibited by all three antibiotics at concentrations >1× MIC; however, none were able to eliminate spores effectively, which were present at the start of the experiment.
Conclusion. The data presented here demonstrate the potent in vitro bactericidal activity of cadazolid against different ribotypes of C. difficile, although on a limited set of strains.
-
-
-
A clinical isolate of Escherichia coli co-harbouring mcr-1 and bla NDM-5 in Japan
More LessThe mcr-1 gene encodes a phosphoethanolamine transferase, which confers resistance to colistin by transferring phosphoethanolamine to lipid A. This study describes the emergence of a colistin- and carbapenem-resistant clinical isolate of Escherichia coli harbouring mcr-1 and bla NDM-5 genes, located on 90 and 150 Kb plasmids, respectively. The isolate belonged to ST132. This is the first report of a clinical isolate in Japan co-harbouring mcr-1 and bla NDM-5.
-
-
-
Antimicrobial resistance profiles of Shigella dysenteriae isolated from travellers returning to the UK, 2004–2017
More LessPurpose. Antimicrobial resistance (AMR) profiles of 754 strains of Shigella dysenteriae isolated between 2004 and 2017 from UK travellers reporting symptoms of gastrointestinal (GI) disease were reviewed to look for evidence of emerging AMR associated with travellers’ diarrhoea.
Methodology. A travel history was provided for 72.7 % (548/754) of cases, of which 90.9 % (498/548) reported travel outside the UK within 7 days of onset of symptoms, and 9.1 % (50/498) reported no travel in that time frame. During the course of this study, whole genome sequencing (WGS) was implemented for GI disease surveillance, and we compared phenotypic AMR profiles with those derived from WGS data (n=133).
Results/Key findings. The phenotypic and genotypic AMR results correlated well, with 90.1 % (121/133) isolates having concordant results to 10 classes of antimicrobials. Resistance to the first-line drugs commonly used in the treatment of shigellosis was observed throughout the study (ampicillin, 54.1%; chloramphenicol, 33.7 %; sulphonamides, 76.0 %; trimethoprim, 80.0%). Between 2004 and 2017, resistance to all classes of antimicrobials (except the phenicols) increased. The proportion of isolates exhibiting reduced susceptibility to ciprofloxacin increased from 3.8 % in 2004 to 75.7 % in 2017, and this was significantly associated with cases reporting travel to Asia compared to Africa (P<0.001). Of the 201 sequenced isolates, 3.0 % (20/201) had either bla CTX-M-15 or bla CMY-4.
Conclusions. Increasing MDR, along with resistance to the fluroquinolones and the third generation cephalosporins, in Shigella dysenteriae causing travellers’ diarrhoea provides further evidence for the need to regulatethe use of antimicrobial agents and continuous monitoring of emerging AMR.
-
-
-
Bactericidal efficacy of molybdenum oxide nanoparticles against antimicrobial-resistant pathogens
More LessMultidrug-resistant bacteria pose a major threat to effective antibiotics and alternatives to fight multidrug-resistant pathogens are needed. We synthetized molybdenum oxide (MoO3) nanoparticles (NP) and determined their antibacterial activity against 39 isolates: (i) eight Staphylococcus aureus, including representatives of methicillin-resistant S. aureus epidemic clones; (ii) six enterococci, including vancomycin-resistant isolates; and (iii) 25 Gram-negative isolates (Escherichia coli, Klebsiella pneumoniae, Pseudomonas aeruginosa, Acinetobacter baumannii, Enterobacter cloacae), including extended spectrum beta-lactamases and carbapenemases producers. All isolates showed a MoO3 NP MIC of 700–800 mg l−1. MoO3 NP produced a clear inhibition zone for S. aureus and all Gram-negative isolates at concentrations ≥25 mg ml−1 and ≥50 mg ml−1 for enterococci. When the NP solutions were adjusted to pH ~7, the biocidal activity was completely abolished. MoO3 NP create an acidic pH and show a universal antimicrobial activity against susceptible and resistant isolates belonging to the most relevant bacterial species responsible for hospital-acquired infections.
-
-
-
Comprehensive screening of antimicrobials to control phytoplasma diseases using an in vitro plant–phytoplasma co-culture system
More LessPhytoplasmas are plant-pathogenic bacteria that infect many important crops and cause serious economic losses worldwide. However, owing to an inability to culture phytoplasmas, screening of antimicrobials on media is difficult. The only antimicrobials being used to control phytoplasmas are tetracycline-class antibiotics. In this study, we developed an accurate and efficient screening method to evaluate the effects of antimicrobials using an in vitro plant–phytoplasma co-culture system. We tested 40 antimicrobials, in addition to tetracycline, and four of these (doxycycline, chloramphenicol, thiamphenicol and rifampicin) decreased the accumulation of ‘Candidatus (Ca.) Phytoplasma asteris'. The phytoplasma was eliminated from infected plants by the application of both tetracycline and rifampicin. We also compared nucleotide sequences of rRNAs and amino acid sequences of proteins targeted by antimicrobials between phytoplasmas and other bacteria. Since antimicrobial target sequences were conserved among various phytoplasma species, the antimicrobials that decreased accumulation of ‘Ca. P. asteris' may also have been effective against other phytoplasma species. These approaches will provide new strategies for phytoplasma disease management.
-
-
-
Epidemic spread of OXA-48 beta-lactamase in Croatia
More LessBranka Bedenić, Mia Slade, Lidija Žele Starčević, Sanda Sardelić, Mirna Vranić-Ladavac, Ana Benčić, Vlasta Zujić Atalić, Maja Bogdan, Marina Bubonja-Šonje, Maja Tomić-Paradžik, Tatjana Tot, Amarela Lukić-Grlić, Domagoj Drenjančević, Dijana Varda-Brkić, Daniela Bandić-Pavlović, Slobodan Mihaljević, Gernot Zarfel, Marija Gužvinec, Rick Conzemius, Ivan Barišić and Arjana Tambić-AndraševicPurpose. A dramatic increase in OXA-48 β-lactamase was observed recently not only in large hospital centres, but also in smaller suburban hospital centres in geographic areas bordering Croatia. The aim of the study was to analyse the epidemiology, the mechanisms of antibiotic resistance and the routes of spread of OXA-48 carbapenemase in Croatia.
Methods. Carbapenemase and other β-lactamase and fluoroquinolone resistance genes were detected by PCR and sequencing. Whole-genome sequencing (WGS) was performed on five representative isolates. The isolates were genotyped by PFGE.
Results. Forty-eight isolates positive for OXA-48, collected from seven hospital centres in Croatia from May 2016 to May 2017, were analysed (40 Klebsiella pneumoniae, 5 Enterobacter cloacae, 2 Escherichia coli and one Citrobacter freundii). Thirty-three isolates were ESBL positive and harboured group 1 CTX-M 1 β-lactamases. In addition to the β-lactam resistance genes detected by PCR (bla SHV-1, bla OXA-48 and bla OXA-1), WGS of five representative isolates revealed the presence of genes encoding aminoglycoside resistance, aadA2 and aph3-Ia, fluoroquinolone resistance determinants aac(6)Ib-c, oqxA and oqxB, the sulfonamide resistance gene sul1, and fosA (fosfomycin resistance). IncL plasmid was found in all isolates. Two K. pneumoniae isolates belonged to ST16, two E. cloacae to ST66 and E. coli to ST354. K. pneumoniae isolates were allocated to five clusters by PFGE which occured in different hospitals, indicating epidemic spread.
Conclusions. The OXA-48-positive organisms found in this study showed wide variability in antibiotic susceptibility, β-lactamase content and PFGE banding patterns. This study revealed a switch from the predominance of VIM-1 in 2012–2013 to that of OXA-48 in the 2015 to 2017.
-
-
-
Genome analysis of methicillin resistance in Macrococcus caseolyticus from dairy cattle in England and Wales
More LessSpecies of the genus Macrococcus are widespread commensals of animals but are becoming increasingly recognised as veterinary pathogens. They can encode methicillin resistance and are implicated in its spread to the closely-related, but more pathogenic, staphylococci. In this study we have identified 33 isolates of methicillin-resistant Macrococcus caseolyticus from bovine bulk tank milk from England and Wales. These isolates were characterised to provide insight into the molecular epidemiology of M. caseolyticus and to discern the genetic basis for their methicillin resistance. Antimicrobial susceptibility testing was performed by Vitek2 and disc diffusion. Isolates were whole-genome sequenced to evaluate phylogenetic relationships and the presence of methicillin resistance determinants, mecA–D. All 33 isolates were phenotypically methicillin-resistant according to cefoxitin disc diffusion, cefoxitin Etest and oxacillin resistance assessed by Vitek2. In contrast only a single isolate was resistant in the Vitek2 cefoxitin screen. Twenty-seven isolates were positive for mecD and six were positive for mecB. mecA and mecC were not detected. The results of phylogenetic analysis indicated that these methicillin-resistant isolates represented a heterogeneous population with both mecB and mecD found in diverse isolates. Isolates had a widespread distribution across the sampled region. Taken together with the role of M. caseolyticus in veterinary infections, including bovine mastitis, and in the potential spread of methicillin resistance to more pathogenic staphylococci, this work highlights the need to better understand their epidemiology and for increased awareness among veterinary microbiology laboratories.
-
-
-
Genomic surveillance of Neisseria gonorrhoeae to investigate the distribution and evolution of antimicrobial-resistance determinants and lineages
More LessThe first extensively drug resistant (XDR) Neisseria gonorrhoeae strain with high resistance to the extended-spectrum cephalosporin ceftriaxone was identified in 2009 in Japan, but no other strain with this antimicrobial-resistance profile has been reported since. However, surveillance to date has been based on phenotypic methods and sequence typing, not genome sequencing. Therefore, little is known about the local population structure at the genomic level, and how resistance determinants and lineages are distributed and evolve. We analysed the whole-genome sequence data and the antimicrobial-susceptibility testing results of 204 strains sampled in a region where the first XDR ceftriaxone-resistant N. gonorrhoeae was isolated, complemented with 67 additional genomes from other time frames and locations within Japan. Strains resistant to ceftriaxone were not found, but we discovered a sequence type (ST)7363 sub-lineage susceptible to ceftriaxone and cefixime in which the mosaic penA allele responsible for reduced susceptibility had reverted to a susceptible allele by recombination. Approximately 85 % of isolates showed resistance to fluoroquinolones (ciprofloxacin) explained by linked amino acid substitutions at positions 91 and 95 of GyrA with 99 % sensitivity and 100 % specificity. Approximately 10 % showed resistance to macrolides (azithromycin), for which genetic determinants are less clear. Furthermore, we revealed different evolutionary paths of the two major lineages: single acquisition of penA X in the ST7363-associated lineage, followed by multiple independent acquisitions of the penA X and XXXIV in the ST1901-associated lineage. Our study provides a detailed picture of the distribution of resistance determinants and disentangles the evolution of the two major lineages spreading worldwide.
-
-
-
Taxonogenomics reveal multiple novel genomospecies associated with clinical isolates of Stenotrophomonas maltophilia
More LessStenotrophomonas maltophilia has evolved as one of the leading multidrug-resistant pathogens responsible for a variety of nosocomial infections especially in highly debilitated patients. As information on the genomic and intraspecies diversity of this clinically important pathogen is limited, we sequenced the whole genome of 27 clinical isolates from hospitalized patients. Phylogenomic analysis along with the genomes of type strains suggested that the clinical isolates are distributed over the Stenotrophomonas maltophilia complex (Smc) within the genus Stenotrophomonas. Further genome-based taxonomy coupled with the genomes of type strains of the genus Stenotrophomonas allowed us to identify five cryptic genomospecies, which are associated with the clinical isolates of S. maltophilia and are potentially novel species. These isolates share a very small core genome that implies a high level of genetic diversity within the isolates. Recombination analysis of core genomes revealed that the impact of recombination is more than mutation in the diversification of clinical S. maltophilia isolates. Distribution analysis of well-characterized antibiotic-resistance and efflux pump genes of S. maltophilia across multiple novel genomospecies provided insights into its antibiotic-resistant ability. This study supports the existence of multiple cryptic species within the Smc besides S. maltophilia, which are associated with human infections, and highlights the importance of genome-based approaches to delineate bacterial species. This data will aid in improving clinical diagnosis and for understanding species-specific clinical manifestations of infection due to Stenotrophomonas species.
-
-
-
Transcriptomic analysis of longitudinal Burkholderia pseudomallei infecting the cystic fibrosis lung
More LessThe melioidosis bacterium, Burkholderia pseudomallei, is increasingly being recognised as a pathogen in patients with cystic fibrosis (CF). We have recently catalogued genome-wide variation of paired, isogenic B. pseudomallei isolates from seven Australasian CF cases, which were collected between 4 and 55 months apart. Here, we extend this investigation by documenting the transcriptomic changes in B. pseudomallei in five cases. Following growth in an artificial CF sputum medium, four of the five paired isolates exhibited significant differential gene expression (DE) that affected between 32 and 792 genes. The greatest number of DE events was observed between the strains from patient CF9, consistent with the hypermutator status of the latter strain, which is deficient in the DNA mismatch repair protein MutS. Two patient isolates harboured duplications that concomitantly increased expression of the β-lactamase-encoding gene penA, and a 35 kb deletion in another abolished expression of 29 genes. Convergent expression profiles in the chronically-adapted isolates identified two significantly downregulated and 17 significantly upregulated loci, including the resistance-nodulation-division (RND) efflux pump BpeEF–OprC, the quorum-sensing hhqABCDE operon, and a cyanide- and pyocyanin-insensitive cytochrome bd quinol oxidase. These convergent pathoadaptations lead to increased expression of pathways that may suppress competing bacterial and fungal pathogens, and that enhance survival in oxygen-restricted environments, the latter of which may render conventional antibiotics less effective in vivo. Treating chronically adapted B. pseudomallei infections with antibiotics designed to target anaerobic infections, such as the nitroimidazole class of antibiotics, may significantly improve pathogen eradication attempts by exploiting this Achilles heel.
-
-
-
Trends in fluoroquinolone resistance in Campylobacter
More LessMembers of the genus Campylobacter remain a leading cause of bacterial gastroenteritis worldwide. Infection is usually self-limiting but in severe cases may require antibiotic treatment. In a recent statement by the World Health Organization (WHO) Campylobacter was named as one of the 12 bacteria that pose the greatest threat to human health because they are resistant to antibiotics. In this mini review we describe recent trends in fluoroquinolone (FQ) (particularly ciprofloxacin) resistance in strains of members of the genus Campylobacter isolated from livestock and clinical samples from several countries. Using evidence from phenotyping surveys and putative resistance prediction from DNA sequence data, we discuss the acquisition and spread of FQ resistance and the role of horizontal gene transfer and describe trends in FQ-resistance in samples from livestock and clinical cases. This review emphasises that FQ resistance remains common among isolates of members of the genus Campylobacter from various sources.
-
-
-
Antifungal susceptibility and virulence of Candida parapsilosis species complex: an overview of their pathogenic potential
More LessRaimunda Sâmia Nogueira Brilhante, Jamille Alencar Sales, Maria Lucilene Queiroz da Silva, Jonathas Sales de Oliveira, Lucas de Alencar Pereira, Waldemiro Aquino Pereira-Neto, Rossana de Aguiar Cordeiro, José Júlio Costa Sidrim, Débora de Souza Collares Maia Castelo-Branco and Marcos Fábio Gadelha RochaPurpose. Antifungal resistance and several putative virulence factors have been associated with the pathogenicity of the Candida parapsilosis species complex. The objective of this study was to evaluate the antifungal susceptibility, the production of virulence factors and the pathogenicity of the C. parapsilosis complex.
Methodology. Overall, 49 isolates of C. parapsilosis sensu stricto, 19 C. orthopsilosis and nine C. metapsilosis were used. The planktonic and biofilm susceptibility to fluconazole, itraconazole, voriconazole, amphotericin B and caspofungin was assessed using a broth microdilution assay. Finally, the production of biofilm and hydrolytic enzymes and the fungal pathogenicity against Caenorhabditis elegans were investigated.
Results/Key findings. Overall, one C. orthopsilosis was resistant to caspofungin and susceptible-dose-dependent to itraconazole, the other two C. orthopsilosis were susceptible-dose-dependent to fluconazole and itraconazole, and one C. metapsilosis was susceptible-dose-dependent to azoles. A total of 67.5 % of the isolates were biofilm producers. Amphotericin B and caspofungin caused the greatest reduction in the metabolic activity and biomass of mature biofilms. Phospholipase and protease production was observed in 55.1 % of C. parapsilosis sensu stricto, 42.1 % of C. orthopsilosis and 33.3 % of C. metapsilosis isolates. Moreover, 57.9 % of C. orthopsilosis and 20.4 % of C. parapsilosis sensu stricto isolates were β-haemolytic, and all C. metapsilosis were α-haemolytic. Finally, the C. parapsilosis complex caused high mortality of C. elegans after 96 h of exposure.
Conclusion. These results reinforce the heterogeneity of these cryptic species for their antifungal susceptibility, virulence and pathogenic potential, emphasizing the relevance of monitoring these emerging pathogens.
-
-
-
Extended-spectrum β-lactamase-producing and carbapenemase-producing Enterobacteriaceae
More LessAntimicrobial resistance (AMR) is a global public-health emergency, which threatens the advances made by modern medical care over the past century. The World Health Organization has recently published a global priority list of antibiotic-resistant bacteria, which includes extended-spectrum β-lactamase-producing Enterobacteriaceae and carbapenemase-producing Enterobacteriaceae. In this review, we highlight the mechanisms of resistance and the genomic epidemiology of these organisms, and the impact of AMR.
-
-
-
Fluoroquinolone resistance in Salmonella: insights by whole-genome sequencing
More LessFluoroquinolone (FQ)-resistant Salmonella spp. were listed by the WHO in 2017 as priority pathogens for which new antibiotics were urgently needed. The overall global burden of Salmonella infections is high, but differs per region. Whereas typhoid fever is most prevalent in South and South-East Asia, non-typhoidal salmonellosis is prevalent across the globe and associated with a mild gastroenteritis. By contrast, invasive non-typhoidal Salmonella cause bloodstream infections associated with high mortality, particularly in sub-Saharan Africa. Most Salmonella strains from clinical sources are resistant to first-line antibiotics, with FQs now being the antibiotic of choice for treatment of invasive Salmonella infections. However, FQ resistance is increasingly being reported in Salmonella, and multiple molecular mechanisms are already described. Whole-genome sequencing (WGS) is becoming more frequently used to analyse bacterial genomes for antibiotic-resistance markers, and to understand the phylogeny of bacteria in relation to their antibiotic-resistance profiles. This mini-review provides an overview of FQ resistance in Salmonella, guided by WGS studies that demonstrate that WGS is a valuable tool for global surveillance.
-
-
-
High diversity in SCCmec elements among multidrug-resistant Staphylococcus haemolyticus strains originating from paediatric patients; characterization of a new composite island
More LessPurpose. Staphylococcus haemolyticus has emerged as a highly antimicrobial-resistant healthcare-associated pathogen, in particular for patients admitted to neonatal intensive care. The objective of this study was to study the nature of SCCmec types among MDR-SH strains isolated from paediatric patients.
Methodology. S. haemolyticus strains (n=60) were isolated from paediatric patients. Antibiotic resistance patterns were established using the disk agar diffusion and micro-broth dilution methods. SCCmec typing was performed using whole-genome sequencing (WGS) and an additional PCR analysis.
Results. All S. haemolyticus isolates demonstrated multidrug resistance. Using WGS, various novel mec types and combinations of SCCmec types were found, including a new composite island [SCCmec type V (Vd)+SCC cad/ars/cop] comprising 30 % of the strains. SCCmec type V was identified in 23 % of the isolates. A combination of the mecA gene enclosed by two copies of IS431 and absence of the mecRI and ccr genes was identified in 11 strains. In total, mecA regulatory genes were absent in all SH isolates used in this study.
Conclusion. A high diversity of SCCmec elements with the prevalence of a new composite island was determined among MRSH strains. The structure of the composite island represented by MDR-SH strains in this study, in combination with the presence of a restriction-modification system type III, is described for the first time in this study. The presence of an 8 bp direct repeat (DR) and the sequences flanking the DR may support the integration of the mecA gene complex as a composite transposon (IS431-mecA-IS431) independently from recombinase genes.
-
-
-
KPC-2 producing ST101 Klebsiella pneumoniae from bloodstream infection in India
More LessThis study characterizes KPC-2 producing Klebsiella pneumoniae belonging to ST101. Whole genome sequencing using the Ion Torrent PGM platform with 400 bp chemistry was performed. bla KPC-2 was found on an IncFIIK plasmid associated with ISKpn6 and ISKpn7 without Tn4401. This is the first report of KPC-2 K. pneumoniae from bacteremia in India. The isolate also coded for other resistance genes such as aadA1, aadA2, armA, aac(3)-Ild, aac(6′)-Ild for aminoglycoside; bla SHV-11 , bla TEM-1B , bla OXA-9, for β-lactams and aac(6′)-Ild, oqxA, oqxB, qnrB1 for fluoroquinolones. It belonged to the K17 capsular type. India is endemic to New Delhi metallo-β-lactamase and OXA48-like carbapenemases and K. pneumoniae carbapenemase (KPC) is seldom reported. With high rates of carbapenem resistance, emergence of KPC in India will challenge patient management. The isolate was susceptible to colistin. The patient had a fatal outcome.
-
-
-
Occurrence and characterization of extended-spectrum cephalosporin-resistant Enterobacteriaceae in healthy household dogs in Greece
More LessExtended-spectrum cephalosporin- and/or carbapenem-resistant (ESCR and/or CarbR) Enterobacteriaceae constitute a public health hazard because of limited treatment options and are endemic among humans in Greece. Recently, ESCR and CarbR Enterobacteriaceae have been increasingly isolated from companion animals, stressing their potential role as a reservoir for humans. However, the presence of ESCR bacteria in companion animals within Greek households has not been determined yet. Genes conferring the ESCR and CarbR phenotype were detected among canine isolates and their chromosomal or plasmid location was determined. Standard methods were applied for plasmid characterization. The clonal relatedness of the recovered isolates was examined by multilocus sequence typing (MLST). Here, we report the first findings on the presence of ESCR Enterobacteriaceae in healthy Greek dogs. ESCR Escherichia coli isolates were associated with different sequence types (STs), including the human pandemic ST131 clone. The occurrence of human-related ESBL/pAmpC genes, plasmid types and/or strain STS in this animal reservoir suggests possible bilateral transmission.
-
-
-
Relevance of antifungal penetration in biofilm-associated resistance of Candida albicans and non-albicans Candida species
More LessThe role of penetration limitation in Candida biofilm-associated antifungal resistance remains unclear. Most of the previous work has been done on Candida albicans, although non-albicans (NAC) species are also implicated in invasive candidiasis and the biofilm matrix has been shown to vary amongst different species. Only a few studies have evaluated clinical isolates. This study aimed to determine the relevance of penetration limitation in the antifungal resistance of biofilms formed by C. albicans and NAC clinical isolates, using an agar disk diffusion assay. The penetration of posaconazole and amphotericin B through the biofilms was significantly reduced. Fluconazole, voriconazole and caspofungin showed a superior penetration capacity in C. albicans, Candida tropicalis and Candida parapsilosis biofilms, but exhibited inter-species and strain/isolate variation. Candida krusei biofilms were the most resilient to antifungal permeation. All of the antifungal drugs failed to kill the biofilm cells, independent of penetration, suggesting that the other factors contribute markedly to the recalcitrance of the biofilms.
-
-
-
Contribution of efflux to colistin heteroresistance in a multidrug resistant Acinetobacter baumannii clinical isolate
More LessPurpose. The mechanisms underlying colistin heteroresistance in Acinetobacter baumannii are not fully understood. Here, we investigated the role of efflux in colistin-heteroresistant populations of a multidrug-resistant (MDR) A. baumannii clinical isolate.
Methodology. Three colistin-resistant A. baumannii strain variants isolated from the same clinical sample were studied for the presence of heteroresistance to colistin by drug susceptibility testing, genotyping and drug resistance target mutation analysis. The existence of active efflux was studied by synergism assays with efflux inhibitors, real-time efflux activity measurements and analysis of the mRNA transcriptional levels of selected efflux pump genes in response to colistin.
Results. All of the strain variants belong to the ST218, clonal complex 92, international clonal lineage II. Different colistin susceptibility levels were observed among the three strain variants, indicating that colistin-heteroresistant subpopulations were being selected upon exposure to colistin. No mutations were found in the genes lpxACD and pmrAB, which are associated with colistin resistance. The results showed the existence of synergistic interactions between efflux inhibitors and colistin and ethidium bromide. Real-time efflux assays demonstrated that the three strain variants had increased efflux activity that could be inhibited in the presence of the inhibitors. The efflux pump genes adeB, adeJ, adeG, craA, amvA, abeS and abeM were found to be overexpressed in the strain variants in response to colistin exposure.
Conclusion. This study shows that efflux activity contributes to colistin heteroresistance in an MDR A. baumannii clinical isolate. The use of efflux inhibitors as adjuvants of the therapy can resensitize A. baumannii to colistin and prevent the emergence of drug resistance.
-
-
-
Genetic analysis of invasive Escherichia coli in Scotland reveals determinants of healthcare-associated versus community-acquired infections
More LessBacteraemia caused by Escherichia coli is a growing problem with a significant mortality. The factors that influence the acquisition and outcome of these infections are not clear. Here, we have linked detailed genetic data from the whole-genome sequencing of 162 bacteraemic isolates collected in Scotland, UK, in 2013–2015, with clinical data in order to delineate bacterial and host factors that influence the acquisition in hospital or the community, outcome and antibiotic resistance. We identified four major sequence types (STs) in these isolates: ST131, ST69, ST73 and ST95. Nearly 50 % of the bacteraemic isolates had a urinary origin. ST69 was genetically distinct from the other STs, with significantly less sharing of accessory genes and with a distinct plasmid population. Virulence genes were widespread and diversely distributed between the dominant STs. ST131 was significantly associated with hospital-associated infections (HAIs), and ST69 with those from the community. However, there was no association of ST with outcome, although patients with HAI had a higher immediate mortality compared to those with community-associated infections (CAIs). Genome-wide association studies revealed genes involved in antibiotic persistence as significantly associated with HAIs and those encoding elements of a type VI secretion system with CAIs. Antibiotic resistance was common, and there were networks of correlated resistance genes and phenotypic antibiotic resistance. This study has revealed the complex interactions between the genotype of E. coli and its ability to cause bacteraemia, and some of the determinants influencing hospital or community acquisition. In part, these are shaped by antibiotic usage, but strain-specific factors are also important.
-
-
-
Identification and prevalence of RND family multidrug efflux pump oqxAB genes in Enterococci isolates from swine manure in China
More LessPurpose. The resistance/nodulation/cell division (RND) family multidrug efflux pump, OqxAB, has been identified as one of the leading mechanisms of plasmid-mediated quinolone resistance and has become increasingly prevalent among Enterobacteriaceae in recent years. However, oqxAB genes have not yet been reported in Enterococcus isolates. The aim of the present study was to identify the oqxAB genes and investigate their prevalence among Enterococcus from swine manure in China.
Methodology. The oqxAB genes were screened in 87 Enterococcus isolates by PCR. The transferability of the oqxAB genes in Enterococcus was determined by conjugation experiments. The genetic environment of oqxAB genes was investigated by cloning experiments, PCR mapping and sequencing.
Results. A high prevalence (86.2 %) of olaquindox resistance was observed in Enterococcus and 98.9 % isolates exhibited multidrug-resistance phenotypes. The occurrence of oqxA and oqxB in Enterococcus was also high (79.3 and 65.5 %, respectively). Sequence analysis of the cloned fragment indicated that the oqxAB cassette was linked to an incomplete Tn5 transposon containing aph(3′)-IIa and flanked by IS26 [IS26-oqxAB-IS26-aph(3′)-IIa]. The oqxAB–aph(3′)-IIa-positive transconjugant or transformant showed resistance or reduced susceptibility to enrofloxacin, ciprofloxacin, olaquindox, mequindox, florfenicol, neomycin and kanamycin.
Conclusion. This is the first time that the oqxAB genes have been identified in Enterococcus faecalis from swine manure. The genetic linkage of oqxAB–aph(3′)-IIa in Enterococcus has not been described before. The high prevalence of oqxAB genes in Enterococcus suggests that it may constitute a reservoir for oqxAB genes and pose a potential threat to public health.
-
-
-
Surveillance of antimicrobial resistance in Neisseria meningitidis strains isolated from invasive cases in Brazil from 2009 to 2016
More LessPurpose. To describe the antimicrobial resistance profile of Neisseria meningitidis isolates causing invasive disease in Brazil from 2009 to 2016.
Methodology. Among 3548 N. meningitidis isolates received, 2888 (81.4 %) were analysed for antimicrobial resistance using the broth microdilution technique, as recommended by the Clinical and Laboratory Standards Institute. Isolates were tested for ciprofloxacin, chloramphenicol, ceftriaxone, penicillin G, ampicillin and rifampin.
Results. All the isolates tested were susceptible to ceftriaxone, while 953 (33.0 %), 1307 (45.3 %) and 2 (0.07 %) isolates were penicillin G-, ampicillin- and rifampin-intermediate, respectively. Resistance to rifampin, ciprofloxacin and chloramphenicol was shown by three isolates (0.1 %), two isolates (0.07 %) and one (0.03 %) isolate, respectively. Although no isolates were resistant to penicillin G in the period of 2009–2016, our results show an upward trend in minimum inhibitory concentrations (MICs) for this drug as of 2010 (P<0.001). There was no significant difference between different gender and age groups of patients for reduced susceptibility to penicillin G. There was a higher frequency of isolates with reduced susceptibility to penicillin G in the South and Southeast regions (P<0.001). This reduced susceptibility was also associated with serotype 19 inside serogroup B (P<0.001).
Conclusion. Despite the decrease in susceptibility to penicillin G and ampicillin observed from 2010, the overall resistance of N. meningitidis isolates to the antimicrobials tested remained uncommon and sporadic, confirming their efficacy for chemoprophylaxis or treatment of invasive meningococcal disease (IMD) in Brazil. Continued surveillance of N. meningitidis antimicrobial susceptibility profiles is important in order to monitor variations in resistance either geographically, over time or in association with emergent clones.
-
-
-
Ceftriaxone-resistant Salmonella Typhi carries an IncI1-ST31 plasmid encoding CTX-M-15
More LessPurpose. Ceftriaxone is the drug of choice for typhoid fever and the emergence of resistant Salmonella Typhi raises major concerns for treatment. There are an increasing number of sporadic reports of ceftriaxone-resistant S. Typhi and limiting the risk of treatment failure in the patient and outbreaks in the community must be prioritized. This study describes the use of whole genome sequencing to guide outbreak identification and case management.
Methodology. An isolate of ceftriaxone-resistant S. Typhi from the blood of a child taken in 2000 at the Popular Diagnostic Center, Dhaka, Bangladesh was subjected to whole genome sequencing, using an Illumina NextSeq 500 and analysis using Geneious software.
Results/Key findings. Comparison with other ceftriaxone-resistant S. Typhi revealed an isolate from the Democratic Republic of the Congo in 2015 as the closest relative but no evidence of an outbreak. A plasmid belonging to incompatibility group I1 (IncI1-ST31) which included bla CTX-M-15 (ceftriaxone resistance) associated with ISEcp-1 was identified. High similarity (90 %) was seen with pS115, an IncI1 plasmid from S. Enteritidis, and with pESBL-EA11, an incI1 plasmid from E. coli (99 %) showing that S. Typhi has access to ceftriaxone resistance through the acquisition of common plasmids.
Conclusions. The transmission of ceftriaxone resistance from E. coli to S. Typhi is of concern because of clinical resistance to ceftriaxone, the main stay of typhoid treatment. Whole genome sequencing, albeit several years after the isolation, demonstrated the success of containment but clinical trials with alternative agents are urgently required.
-
-
-
Occurrence of IMP-1 in non-baumannii Acinetobacter clinical isolates from Brazil
More LessThe aim of this study was to characterize the presence of carbapenemase-encoding genes in distinct species of Acinetobacter spp. isolated from Brazilian hospitals. Five carbapenem-resistant Acinetobacter spp. isolates (two Acinetobacter pittii, two Acinetobacter bereziniae and one Acinetobacter junii) recovered from two distinct hospitals between 2000 and 2016 were included in this study. All of the isolates harboured bla IMP-1, which was inserted into In86, a class 1 integron. Pulsed field gel eletrophoresis analysis showed that both A. pittii were identical, while the two A. berezinae isolates were considered to be clonally related. In this study, we demonstrated that mobile elements carrying carbapenemase-encoding genes such as In86 may persist for a long period, allowing their mobilization from A. baumannii to other Acinetobacter spp. that are usually susceptible to multiple antimicrobials.
-
-
-
Retrospective study on clonal relationship of multidrug-resistant Klebsiella spp. indicates closed circulation and initiation of clonal divergence
More LessPurpose. Antibiotic resistance patterns often exhibit geographical variations. Periodic analyses of resistance spectra and phylogenetic trends are important guides for facilitating judicious use of therapeutic interventions. The present study retrospectively analysed the infection trends, resistance patterns, and clonal relationships between isolates of Klebsiella spp. from a tertiary care hospital.
Methodology. Bacterial isolates were collected from January 2013 to June 2014 and their resistance profiles were identified using an automated bacterial identification system. A phylogenetic tree was constructed using housekeeping genes with Molecular Evolutionary Genetic Analysis software. The d N/d S ratio was determined by the Synonymous Non-synonymous Analysis Program while polymorphic sites, and the difference per site was calculated using DNA Sequence Polymorphism software. Statistical Package for Social Science software was used to perform all statistical analyses.
Key findings. The results of this study indicated the prevalence of community-acquired urinary tract and lower respiratory tract infections caused by Klebsiella spp. among geriatric patients. The occurrence of new allelic profiles, a low d N/d S ratio and the lack of strong evolutionary descent between isolates indicated that mutations play a major role in the evolution of the organism.
Conclusion. The findings of this study highlight the consequences of antimicrobial agents exerting a silent and strong selective force on the evolution of Klebsiella spp. The expansion of such analyses is of great importance for addressing rapidly emerging antibiotic-resistant opportunistic pathogens.
-
-
-
Antimicrobial activity against Mycobacterium tuberculosis under in vitro lipid-rich dormancy conditions
More LessAlthough tuberculosis treatment is dependent on drug-susceptibility testing (DST) and molecular drug-resistance detection, treatment failure and relapse remain a challenge. This could be partially due to the emergence of antibiotic-tolerant dormant mycobacteria, where host lipids have been shown to play an important role. This study evaluated the susceptibility of Mycobacterium tuberculosis to two antibiotic combinations – rifampicin, moxifloxacin, amikacin and metronidazole (RIF-MXF-AMK-MTZ), and rifampicin, moxifloxacin, amikacin and pretomanid (RIF-MXF-AMK-PA) – in a lipid-rich dormancy model. Although their effectiveness in in vitro cultures with dextrose as a carbon source has been proved, we observed that none of the antibiotic mixtures were bactericidal in the presence of lipids. The presence of lipids may confer tolerance to M. tuberculosis against the mixture of antibiotics tested and such tolerance could be even higher during the dormant stages. The implementation of lipids in DST on clinical isolates could potentially lead to a better treatment strategy.
-
-
-
Emergence of resistance to ciprofloxacin in Neisseria meningitidis in Brazil
More LessTo prevent secondary invasive meningococcal disease (IMD) cases and outbreaks, antimicrobial prophylaxis of high-risk contacts is indicated. This study reports two ciprofloxacin-resistant Neisseria meningitidis strains in Brazil. The 3523 N. meningitidis isolates collected throughout Brazil from 2009 to 2016 were evaluated for antimicrobial resistance. Meningococcal isolates showing minimal inhibitory concentrations, MICs≥0.125µg ml−1 to ciprofloxacin, were analysed to determine the presence of mutations in the quinolone resistance-determining regions (QRDRs) of gyrA and parC genes. Two ciprofloxacin-resistant N. meningitidis isolates were found, both presenting a single mutation in the quinolone resistance-determining region of the gyrA gene. These results confirmed that ciprofloxacin is still a first-line drug for chemoprophylaxis. However, we highlight the importance of continued surveillance to monitor the trends of N. meningitidis susceptibility profiles to the antimicrobials recommended for chemoprophylaxis and IMD treatment.
-
-
-
Evolution of carbapenem resistance in Acinetobacter baumannii during a prolonged infection
More LessAcinetobacter baumannii is a common causative agent of hospital-acquired infections and a leading cause of infection in burns patients. Carbapenem-resistant A. baumannii is considered a major public-health threat and has been identified by the World Health Organization as the top priority organism requiring new antimicrobials. The most common mechanism for carbapenem resistance in A. baumannii is via horizontal acquisition of carbapenemase genes. In this study, we sampled 20 A. baumannii isolates from a patient with extensive burns, and characterized the evolution of carbapenem resistance over a 45 day period via Illumina and Oxford Nanopore sequencing. All isolates were multidrug resistant, carrying two genomic islands that harboured several antibiotic-resistance genes. Most isolates were genetically identical and represented a single founder genotype. We identified three novel non-synonymous substitutions associated with meropenem resistance: F136L and G288S in AdeB (part of the AdeABC efflux pump) associated with an increase in meropenem MIC to ≥8 µg ml−1; and A515V in FtsI (PBP3, a penicillin-binding protein) associated with a further increase in MIC to 32 µg ml−1. Structural modelling of AdeB and FtsI showed that these mutations affected their drug-binding sites and revealed mechanisms for meropenem resistance. Notably, one of the adeB mutations arose prior to meropenem therapy but following ciprofloxacin therapy, suggesting exposure to one drug whose resistance is mediated by the efflux pump can induce collateral resistance to other drugs to which the bacterium has not yet been exposed.
-